Nutzenbewertung für Arzneimittel
Neue Regeln für Arzneimittelbewertung: Was die HTA-Verordnung für Unternehmen bedeutet
Seit dem 12.01.2025 gilt in der Europäischen Union eine neue Verordnung zur Bewertung von Arzneimitteln und bestimmten Medizinprodukten: Die HTA-Verordnung soll die Klinische Bewertung europaweit harmonisieren und doppelten Aufwand vermeiden. Anstatt einzelnen Nutzenbewertungen für jedes Land, wird es in Zukunft ein einheitliches, europaweit gültiges Verfahren geben.
Die Verordnung (EU) 2021/2282 über die Bewertung von Gesundheitstechnologien (HTA-Verordnung) betrifft die Klinische Bewertung neuer Medikamente und Medizinprodukte. Doch verändert sie die bestehenden Verfahren grundlegend, oder bleibt für Deutschland vieles beim Alten? Welche Folgen hat die Reform für das Verfahren des Arzneimittelmarktneuordnungsgesetzes (AMNOG) und die Pharmaunternehmen hierzulande? Antworten darauf geben die Expertinnen Sarah Schmitter, Director Health Technology Assessment & Outcomes Research bei Pfizer sowie Dr. Eva-Maria Reuter, Senior HTA Manager & Medical Writer bei Advanced Medical Services (AMS).
Was ist die Nutzenbewertung – und warum ist sie wichtig?
Bevor ein neues Medikament auf den Markt kommt, muss es zugelassen werden. Die Zulassung stellt jedoch nur sicher, dass ein Medikament sicher und wirksam ist. Ob es für Patientinnen und Patienten auch tatsächlich einen Mehrwert gegenüber bestehenden Therapien bietet, bewertet dagegen eine Nutzenbewertung, das Health Technology Assessment (HTA). Indirekt hat dies unter anderem auch Auswirkungen auf die Frage, ob ein Medikament von den Krankenkassen erstattet wird, und unter welchen Bedingungen es in das jeweilige Gesundheitssystem aufgenommen wird. „HTA ist die wissenschaftliche Analyse Klinischer Evidenz zu den relativen Auswirkungen einer Gesundheitstechnologie“, erklärt Reuter. „Das HTA ist die Basis für den Zugang zum nationalen Erstattungssystem.“
Was ändert sich mit der HTA-Verordnung?
Die HTA-Verordnung soll vor allem die Klinische Bewertung zentralisieren. Die bisherigen Bewertungen auf nationaler Ebene haben teils zu Verzögerungen und oft doppeltem Aufwand für Unternehmen geführt. Ab 2025 wird die Klinische Nutzenbewertung daher zentral auf EU-Ebene durchgeführt – die Mitgliedstaaten müssen die Ergebnisse dieser Joint Clinical Assessments (JCA) in ihren nationalen Entscheidungen berücksichtigen. Die Preisverhandlungen und die endgültige Entscheidung über die Erstattung bleiben allerdings in nationaler Hand.
Eingeführt wird die HTA-VO schrittweise, und zwar mit folgendem Zeitplan:
- 12.01.2025: Start der EU-weiten Klinischen Bewertungen (Joint Clinical Assessments, JCA) für neuartige Therapien (ATMPs) und onkologische Arzneimittel
- 2026: Bestimmte Hochrisiko-Medizinprodukte
- 2028: Ausweitung auf Orphan Drugs
- 2030: Alle zentral zugelassenen Arzneimittel
 Die HTA-VO wird schrittweise ausgerollt.
Die HTA-VO wird schrittweise ausgerollt.
Quelle: Auszug aus Factsheet - Implementing the EU Health Technology Assessment Regulation (https://health.ec.europa.eu/publications/factsheet-joint-clinical-assessment-medicinal-products-january-2025_en), EU-Kommission; (Creative Commons Attribution 4.0 International licence (https://creativecommons.org/licenses/by/4.0/deed.en)). WEB: ISBN 978-92-68-21595-1
Wie passt das AMNOG zur HTA-Verordnung?
Für Deutschland ist HTA nichts Neues. Hierzulande werden entsprechende Bewertungen bereits seit Jahren durchgeführt, allerdings läuft das Ganze im Rahmen von AMNOG.
Deutschland hat im Rahmen des Arzneimittelmarktneuordnungsgesetzes (AMNOG) bereits jetzt ein einschlägiges Bewertungsverfahren. Seit 2011 müssen Hersteller den Zusatznutzen neuer Medikamente in einem festgelegten Verfahren nachweisen. Das AMNOG-Verfahren besteht aus zwei zentralen Schritten: Zunächst bewerten das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) oder der Gemeinsame Bundesausschuss (G-BA) den Zusatznutzen auf Basis eines Dossiers des Herstellers. Anschließend verhandeln die Hersteller und der Spitzenverband Bund der Krankenkassen (GKV-SV) über den Erstattungspreis.
Die HTA-VO ändert diesen Ablauf nur in einem Punkt: Die Bewertung der Klinischen Studien wird künftig auf europäischer Ebene durchgeführt. Der G-BA nutzt die Ergebnisse der Joint Clinical Assessments (JCA) als Basis für seine nationale Entscheidung, kann aber zusätzliche Analysen verlangen.
Was ändert sich für deutsche Unternehmen?
Für Pharmaunternehmen in Deutschland bleibt der Marktzugang daher weitgehend unverändert. Das AMNOG-Verfahren gibt es weiterhin, ebenso die Preisverhandlungen mit den Krankenkassen. Schmitter schätzt die Lage für deutsche Unternehmen so ein: „Durch die EU HTA-VO wird lediglich ein Teil des Prozesses der Nutzenbewertung auf europäische Ebene verlagert. Der Beschluss zur Nutzenbewertung verbleibt beim Gemeinsamen Bundesausschuss sowie die Verhandlungen zum Erstattungsbetrag verbleiben unverändert in Deutschland.“
Wie läuft das Verfahren ab 2025 ab?
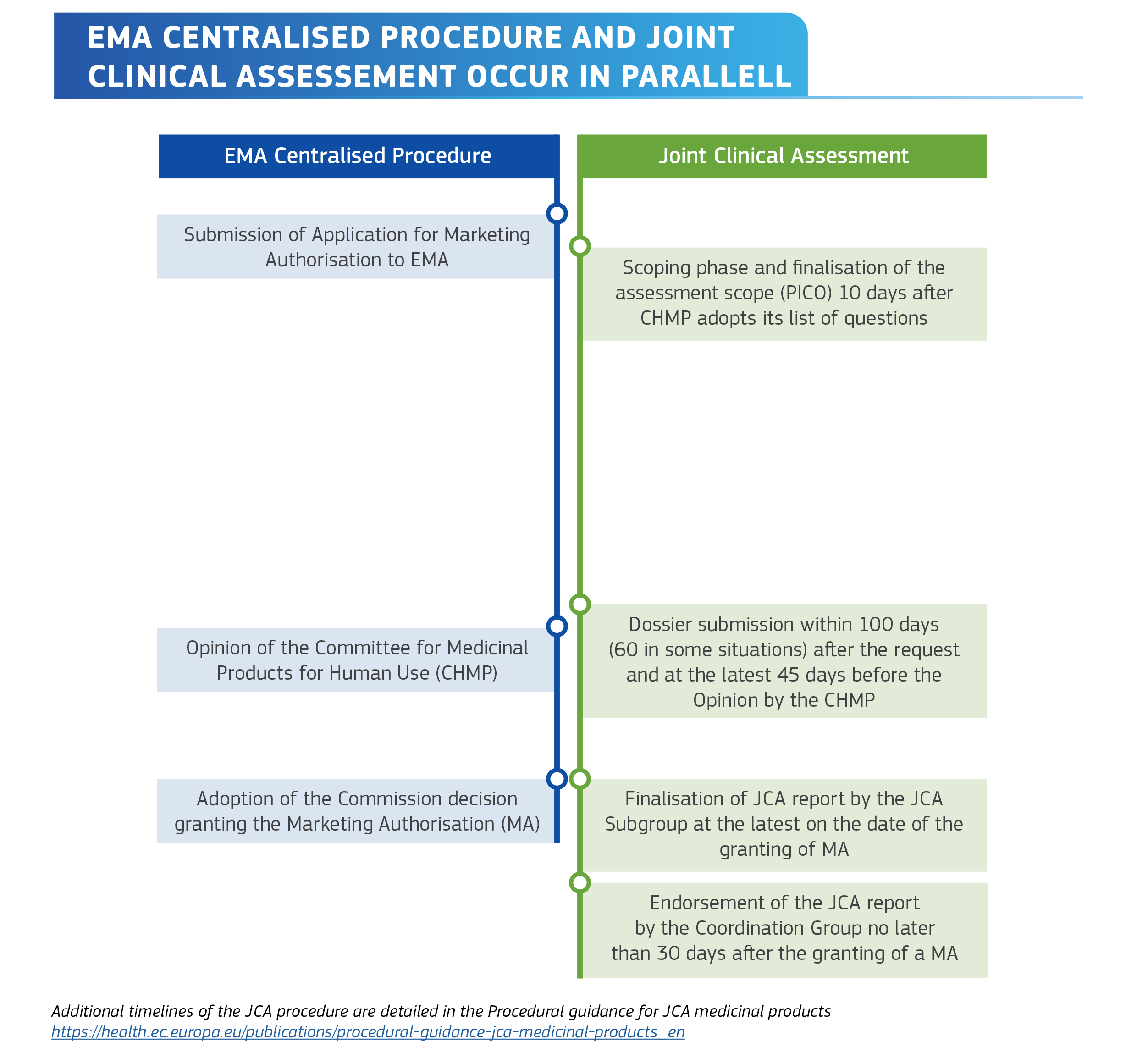 Der Ablauf des HTA-Prozesses parallel zur EMA-Prozedur.
Der Ablauf des HTA-Prozesses parallel zur EMA-Prozedur.
Quelle: Auszug aus Factsheet - Implementing the EU Health Technology Assessment Regulation (https://health.ec.europa.eu/publications/factsheet-joint-clinical-assessment-medicinal-products-january-2025_en), EU-Kommission (Creative Commons Attribution 4.0 International licence (https://creativecommons.org/licenses/by/4.0/deed.en)). WEB: ISBN 978-92-68-21595-1Laut den Expertinnen bedeutet das neue HTA-System allerdings für Unternehmen mitunter straffere Fristen und neue methodische Anforderungen. Der EU HTA-Prozess beginnt deutlich früher als viele bisher übliche nationale Nutzenbewertungen. Unternehmen müssen sich bereits in der Klinischen Studienphase mit den HTA-Anforderungen auseinandersetzen, da die EU-Bewertung strenge methodische Vorgaben hat.
Unternehmen müssen ein HTA-Dossier einreichen, das sämtliche Klinische Studiendaten, Analysen und Nachweise enthält, die für die Bewertung der Klinischen Evidenz erforderlich sind. Nach Einreichen des Dossiers beginnt die Klinische Bewertung durch die zuständigen HTA-Expertinnen und -Experten. Diese analysieren die Wirksamkeit des Arzneimittels im Vergleich zur Standardtherapie. Die Analyse dauert mehrere Monate und endet mit dem Erstellen eines gemeinsamen Klinischen Bewertungsberichts (JCA-Report). Dieser Bericht ist öffentlich zugänglich und dient als Grundlage für nationale HTA-Entscheidungen.
Sobald die gemeinsame Klinische Bewertung abgeschlossen ist, folgt die nationale Phase. Hier entscheidet jeder Mitgliedstaat individuell, wie er den Bericht nutzt, und welche zusätzlichen Analysen er benötigt. In Deutschland übernimmt der Gemeinsame Bundesausschuss (G-BA) die nationale Bewertung und entscheidet über den Zusatznutzen des Arzneimittels.
Was die Expertinnen Unternehmen raten
Auch wenn in Deutschland vieles gleich bleiben wird, bringt die HTA-Verordnung neue Anforderungen mit sich. „Wir empfehlen, sich frühzeitig über den gesamten Prozess zu informieren und sich einen klaren Überblick über die allgemeinen Anforderungen zu verschaffen“, sagt Reuter. „Anschließend sollte man den Prozess für ein spezifisches Produkt im Detail durchgehen, also die einzelnen Arbeitsschritte, Abläufe und Verantwortlichkeiten festlegen.“ Schmitter sieht dabei eine besondere Herausforderung in der internen Organisation der Unternehmen: „Es ist ein neuer und zugleich wichtiger Prozess, den noch kein Arzneimittel durchlaufen hat. Für die Unternehmen bedeutet es, dass sie sich überlegen, wie sie den Prozess, insbesondere Erstellung des EU-Dossiers, intern organisieren.“
Ein zentraler Punkt sei auch die Wahl der Vergleichstherapien. „Im Rahmen des EU HTA wird teilweise erstmalig spezifische Evidenz für die HTA-Anforderungen einzelner Länder vorgelegt werden müssen“, sagt Reuter. Das könne für einige Märkte eine erhebliche Umstellung bedeuten. Um Fehler zu vermeiden, rät sie, die sogenannten Joint Scientific Consultations zu nutzen: „Man sollte die HTA auch so früh wie möglich mitdenken und sich am besten entsprechend beraten lassen.“
Fazit: Großer Wandel für die EU – wenig Änderungen in Deutschland
Die EU HTA-Verordnung könnte den Marktzugang für Medikamente und Medizinprodukte langfristig vereinfachen. Für viele europäische Unternehmen wird die Reform jedoch erst einmal einen organisatorischen Mehraufwand bedeuten. Wer sich frühzeitig vorbereitet und interne Prozesse anpasst, kann profitieren. Deutsche Unternehmen dürfen sich allerdings freuen: Für sie ändert sich letztlich dank des bekannten AMNOG-Prozesses nicht besonders viel.