BIOPRO Spezial: Die Klinische Prüfung im Blick
Zulassung nach MDR - Wann ist eine Klinische Prüfung notwendig?
Seit der Veröffentlichung der MEDDEV 2.7/1 Revision 4 im Jahr 2016 ist der Stellenwert der Klinischen Evidenz innerhalb des CE-Konformitätsbewertungsverfahrens immer weiter in den Vordergrund gerückt. Die Verordnung (EU) 2017/745, die 2017 in Kraft getreten und ab Mai 2021 verpflichtend anzuwenden ist, verschärft diesen Trend. Sie legt einen deutlichen Fokus auf die Klinische Evidenz von Medizinprodukten vor und nach der Markteinführung. Somit steigt die Notwendigkeit Klinischer Prüfungen und Klinischer Studien nach dem Inverkehrbringen.
Die Klinische Prüfung stellt kleine und mittelständische Medizintechnik-Unternehmen und Start-ups, aber auch große, etablierte Medizinproduktehersteller, vor administrative, logistische und finanzielle Herausforderungen.
Innerhalb der Verordnung (EU) 2017/745 (Medical Device Regulation, MDR) und in der ISO 14155 wird hauptsächlich der Begriff „Klinische Prüfung“ verwendet, dieser soll auch vorrangig in diesem Leitfaden benutzt werden. „Klinische Prüfung“ (Clinical Investigation) ist gleichbedeutend mit den Begriffen „Klinischer Versuch“ (Clinical Trial) oder „Klinische Studie“ (Clinical Study). Die Klinische Prüfung bezeichnet eine systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer (Patienten oder Probanden) einbezogen werden, und die zwecks Bewertung der Sicherheit und/oder der Leistung eines Medizinprodukts durchgeführt wird.1,2 Innerhalb der Klinischen Prüfung werden klinische Daten zur Sicherheit oder Leistung im Rahmen der Anwendung eines Medizinprodukts gewonnen. Diese klinischen Anwendungsdaten sind somit eine wichtige Quelle, um innerhalb der Konformitätsbewertung darzulegen, dass das betreffende Medizinprodukt die grundlegenden Sicherheits- und Leistungsanforderungen der MDR erfüllt.3
Um die Sicherheit und Leistung eines Medizinprodukts, einschließlich des klinischen Nutzens, nachzuweisen, können neben Daten aus einer Klinischen Prüfung des betreffenden Medizinprodukts oder eines Äquivalenzprodukts (gleichartiges Medizinprodukt) auch folgende Quellen herangezogen werden:
- wissenschaftliche, klinische Fachliteratur,
- Berichte über sonstige klinische Erfahrungen,
- klinisch relevante Angaben aus der Überwachung nach dem Inverkehrbringen, insbesondere aus der klinischen Nachbeobachtung nach dem Inverkehrbringen (PMCF-Maßnahmen).1
Im Rahmen der Konformitätsbewertung wird festgestellt, ob die Anforderungen der MDR an das Produkt erfüllt worden sind. Dafür werden innerhalb einer Klinischen Bewertung kontinuierlich klinische Daten zu dem betreffenden oder gleichartigen Medizinprodukt generiert, gesammelt, analysiert und bewertet. Die Klinische Bewertung kann sich dabei auf eine Zusammenstellung der derzeit verfügbaren wissenschaftlichen Literatur (Literatur- oder Äquivalenzroute) stützen oder auf die Daten der Klinischen Prüfung des betreffenden Produkts.
Als Voraussetzung für die Äquivalenzroute muss ein gleichartiges Produkt zur Verfügung stehen, bei dem ein ausreichender Klinischer Nachweis (Klinische Evidenz) zur Sicherheit und Leistung in der wissenschaftlichen Literatur dokumentiert ist. Die Gleichartigkeit (biologisch, technisch, klinisch) mit dem betreffenden Produkt muss in der Klinischen Bewertung nachgewiesen werden.
Durch die MDR sind jedoch die Anforderungen zum Nachweis der Gleichartigkeit sehr hoch und stellen eine Hürde bei der Äquivalenzroute dar. Die Verordnung sieht in diesem Zusammenhang explizit vor, dass im Fall von implantierbaren Produkten und Produkten der Klasse III Klinische Prüfungen durchgeführt werden müssen, es sei denn
- das betreffende Produkt wurde durch Änderungen eines bereits von demselben Hersteller in Verkehr gebrachten Produkts konzipiert,
- der Hersteller hat nachgewiesen, dass das geänderte Produkt dem in Verkehr gebrachten Produkt gleichartig und dieser Nachweis von der Benannten Stelle bestätigt worden ist,
- die Klinische Bewertung des in Verkehr gebrachten Produkts reicht aus, um nachzuweisen, dass das geänderte Produkt die einschlägigen Sicherheits- und Leistungsanforderungen erfüllt.4 [Auszug aus der MDR, Artikel 61]
Um die grundlegenden Sicherheits- und Leistungsanforderungen zu erfüllen, muss der Umfang der Klinischen Evidenz (laut MDR „Klinischer Nachweis“) spezifiziert und begründet werden. Dabei soll der Umfang den Merkmalen des Medizinprodukts sowie dessen Zweckbestimmung angemessen sein. Erachtet der Hersteller einen Nachweis auf Grundlage klinischer Daten als ungeeignet, muss er begründen, wie der Nachweis allein aufgrund von Ergebnissen nicht-klinischer Tests, einschließlich Leistungsbewertung, technischer Prüfung („Bench testing“) und vorklinischer Bewertung geführt werden kann.4
Für Medizinproduktehersteller ist es somit eine herausfordernde und essenzielle Aufgabe zu analysieren, in welchem Umfang klinische Daten benötigt werden, und ob eine Klinische Prüfung vor der Markteinführung regulatorisch notwendig ist, um mögliche Evidenzlücken zu schließen.
Folgende Leitfragen können dem Hersteller bei der Einschätzung helfen, ob ein Medizinprodukt im Rahmen der CE- Konformitätsbewertung eine Klinische Prüfung durchlaufen muss:
- Sind bereits Klinische Studien, Publikationen (Klinische Evidenz) zur angewendeten Methodik (Stand der Technik) und dem Produkt bzw. einem gleichartigen Produkt vorhanden?
- In welche Risikoklasse wird das Medizinprodukt eingeordnet, und wie hoch ist der Innovationsgrad?
- Bestehen mögliche Restrisiken oder andere Unsicherheiten bei dem Medizinprodukt (Risikomanagement)?
Dieser Sachverhalt lässt sich in einem „magischen Dreieck“ veranschaulichen. Dabei stellt die Pfeillänge jeweils den Grad der Evidenz, Innovation bzw. des Risikos für das betrachtete Medizinprodukt dar.
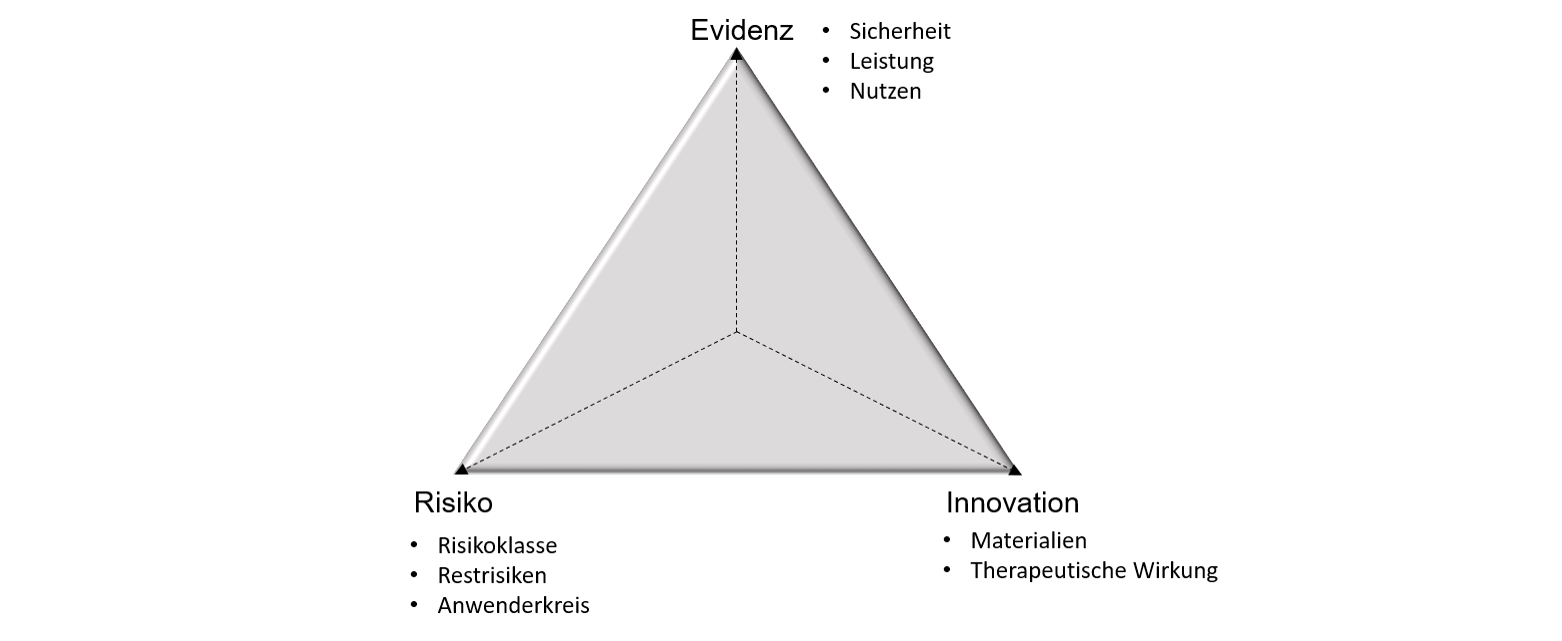 „Magisches Dreieck“ aus Evidenz, Risiko und Innovation als wichtige Parameter für die Einschätzung, ob eine Klinische Prüfung im Rahmen der CE-Konformitätsbewertung benötigt wird. © Robert Radloff
„Magisches Dreieck“ aus Evidenz, Risiko und Innovation als wichtige Parameter für die Einschätzung, ob eine Klinische Prüfung im Rahmen der CE-Konformitätsbewertung benötigt wird. © Robert RadloffEin Produkt, bei dem ein gleichartiges Medizinprodukt der Klasse I (geringes Risiko) bereits seit Jahren am Markt etabliert (geringer Innovationsgrad) und die Klinische Evidenz ausreichend vorhanden ist, kann entsprechend mit hoher Wahrscheinlichkeit über die Äquivalenzroute zugelassen werden (grünes Dreieck).
Handelt es sich dagegen beispielsweise um ein innovatives implantierbares Medizinprodukt (hoher Innovationsgrad) der Klasse III (hohes Risiko), bei dem noch keine ausreichende Klinische Evidenz vorhanden ist, wird mit hoher Wahrscheinlichkeit eine Klinische Prüfung durchgeführt werden müssen, um das Produkt in den Markt einzuführen (rotes Dreieck).
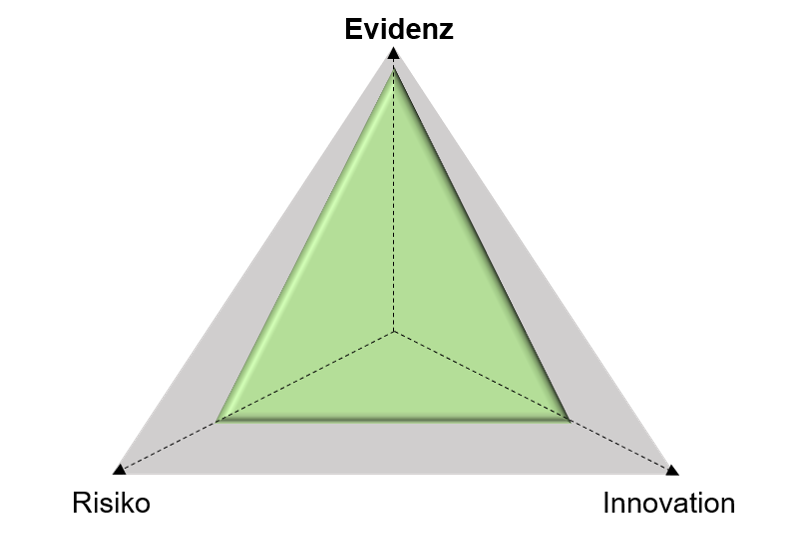 „Magisches Dreieck“, bei dem ein Medizinprodukt ein ausreichendes Maß an Evidenz aufweist in Bezug auf Risiko und Innovationsgrad
© Robert Radloff
„Magisches Dreieck“, bei dem ein Medizinprodukt ein ausreichendes Maß an Evidenz aufweist in Bezug auf Risiko und Innovationsgrad
© Robert Radloff
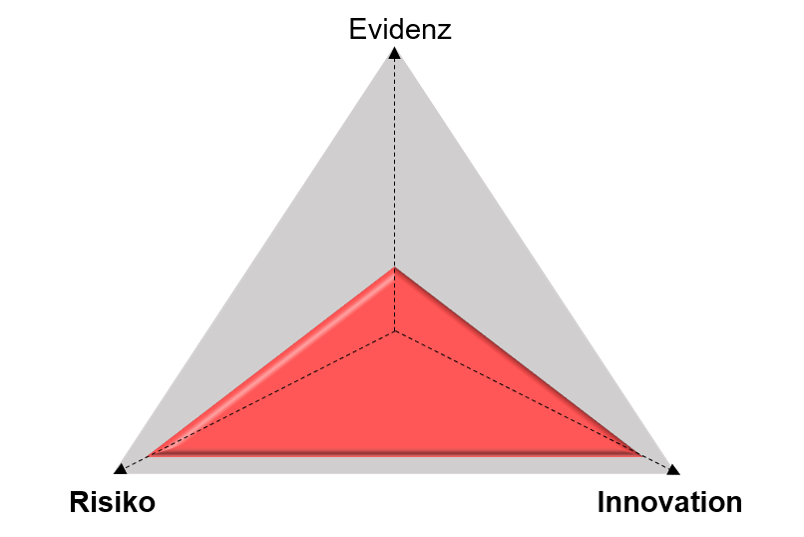 „Magisches Dreieck“, bei dem ein Medizinprodukt eine Evidenzlücke und ein hohes Maß an Risiko und an Innovation aufweist.
© Robert Radloff
„Magisches Dreieck“, bei dem ein Medizinprodukt eine Evidenzlücke und ein hohes Maß an Risiko und an Innovation aufweist.
© Robert Radloff
Es müssen also drei Hauptfaktoren betrachtet werden: Erstens, wie hoch das Risiko des neuen Medizinprodukts ist, zweitens, wie hoch der Innovationsgrad bzw. die Unsicherheit in dem Anwendungsgebiet ist und drittens, ob es ausreichend Klinische Evidenz gibt, die belegt, dass das Produkt sicher und leistungsfähig ist.
Hersteller von Medizinprodukten müssen sich mit der Frage auseinandersetzen, was ausreichend Klinische Evidenz für ihr Medizinprodukt bedeutet. Wann ist nachgewiesen, dass ein Produkt sicher und leistungsfähig ist? Diese Frage wird im ersten Schritt des Plans für die Klinische Bewertung (PKB - engl.: Clinical Evaluation Plan) analysiert.
Der Plan für die Klinische Prüfung muss nach MDR folgende Punkte enthalten (Stand Februar 2021):
- Bestimmung der grundlegenden Sicherheits- und Leistungsanforderungen, die mit relevanten klinischen Daten zu untermauern sind,
- Spezifizierung der Zweckbestimmung des Produkts,
- genaue Spezifizierung der vorgesehenen Zielgruppen mit klaren Indikationen und Kontraindikationen,
- detaillierte Beschreibung des angestrebten klinischen Nutzens für die Patienten mit relevanten konkreten Parametern für das klinische Ergebnis,
- Spezifizierung der für die Prüfung der qualitativen und quantitativen Aspekte der klinischen Sicherheit anzuwendenden Methoden unter deutlicher Bezugnahme auf die Bestimmung der Restrisiken und Nebenwirkungen,
- Nicht erschöpfende Liste und Spezifizierung der Parameter zur auf dem neuesten medizinischen Kenntnisstand beruhenden Bestimmung der Vertretbarkeit des Nutzen-Risiko-Verhältnisses für die verschiedenen Indikationen und die Zweckbestimmung(en) des Produkts,
- Angabe, wie Fragen hinsichtlich des Nutzen-Risiko-Verhältnisses für bestimmte Komponenten wie die Verwendung pharmazeutischer, nicht lebensfähiger tierischer oder menschlicher Gewebe zu klären sind und klinischer Entwicklungsplan: von explorativen Studien, wie Studien zur Erstanwendung am Menschen („First-in-man“-Studien), Durchführbarkeitsstudien und Pilotstudien bis hin zu Bestätigungsstudien, wie pivotale Klinische Prüfungen, und einer klinischen Überwachung nach dem Inverkehrbringen gemäß Teil B dieses Anhangs, unter Angabe von Etappenzielen und Beschreibung möglicher Akzeptanzkriterien.5 [Auszug aus der MDR, Anhang XIV, Teil A 1a]
Die MDR setzt die Hürden bei implantierbaren Produkten und Klasse III-Produkten deutlich höher. Hier muss in den meisten Fällen trotz ausreichender Klinischer Evidenz von einem gleichartigen Produkt eines anderen Herstellers eine Klinische Prüfung durchgeführt werden. Darauf kann nur verzichtet werden, wenn zwischen beiden Herstellern ein Vertrag geschlossen wurde, welcher dem Hersteller des zweiten Produkts ausdrücklich uneingeschränkten Zugang zur technischen Dokumentation gewährt.4
Die Ausführungen zeigen, dass es keine pauschale Antwort auf die Frage, „Klinische Prüfung: Ja oder Nein?“, geben kann, da dies von vielen Einzelfaktoren abhängig ist. Die Entscheidung, ob eine Klinische Prüfung durchgeführt wird, oder die Äquivalenzroute gewählt werden kann, sollte zusammen mit der Benannten Stelle abgestimmt werden. Bei weiterem Klärungsbedarf bietet die zuständige Bundesoberbehörde, in Deutschland das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), eine wissenschaftliche und verfahrenstechnische Beratung an.
Um einen tieferen Einblick in das Thema Klinische Evidenz zu bekommen, empfiehlt sich ein Blick in das MDCG Guidance Dokument 2020-6.6 Dieser Leitfaden beschreibt qualitative und quantitative Merkmale und eine Hierarchie der Klinischen Evidenz, die zur Bestätigung der Konformität gemäß der MDR herangezogen werden kann (MDCG 2020-6 Anhang III).
Zusammenfassung
Mit dem Inkrafttreten der Europäischen Medizinprodukte-Verordnung steigen die Erwartungen, die Sicherheit und Leistung von Produkten mit ausreichender Klinischer Evidenz zu belegen. Anhand der klinischen Daten, die entweder in einer Klinischen Prüfung erhoben oder innerhalb der Äquivalenzroute identifiziert werden, müssen die Hersteller zusammenfassend folgendes nachweisen:
- Erfüllung der grundlegenden Sicherheits- und Leistungsanforderungen,
- den klinischen Nutzen für die Patienten mit relevanten konkreten Parametern für das klinische Ergebnis,
- qualitative und quantitative Aspekte der klinischen Sicherheit,
- Bestimmung der Restrisiken und Nebenwirkungen,
- Vertretbarkeit des Nutzen-Risiko-Verhältnisses.
Besonders für innovative Medizinprodukte höherer Risikoklassen sind eigene klinische Daten unerlässlich geworden. Aus diesem Grund werden zunehmend Klinische Prüfungen und PMCF-Studien verbindlich gefordert, um ein Medizinprodukt zuzulassen bzw. zu rezertifizieren. Hersteller müssen sich also mehr und mehr mit der komplexen Frage auseinandersetzen, was eine ausreichende Klinische Evidenz für ihr Medizinprodukt bedeutet, und wie sie die Anforderungen innerhalb von Klinischen Prüfungen und PMCF-Studien pragmatisch und zielgerichtet umsetzen können.
Wir machen ausdrücklich darauf aufmerksam, dass unser Webangebot lediglich dem unverbindlichen Informationszweck dient. Alle angebotenen Informationen sind ohne Gewähr und erheben keinen Anspruch auf Vollständigkeit und Richtigkeit. Eine Haftung für die Angaben sowie für deren Rechtsverbindlichkeit wird nicht übernommen.