BIOPRO Spezial: Materialänderung von Medizinprodukten
Materialänderung, Nachhaltigkeit und biologische Sicherheit von Medizinprodukten – ein Ausflug in den regulatorischen Dschungel
Seit des Inkrafttretens der Medizinprodukteverordnung (MDR) im Jahre 2017 hat sich einiges getan. Für manche Unternehmen stellen die MDR-Anforderungen weiterhin Probleme dar, während sich andere schon mit der Umsetzung zahlreicher weiterer Richtlinien und Verordnungen beschäftigen, die sich aufgrund der MDR ergeben haben. Neben diesen Regularien gilt es heutzutage, noch weitere Dinge wie z. B. Nachhaltigkeit zu beachten. Gerade im Hinblick auf potenzielle oder notwendige Materialänderungen von Medizinprodukten müssen in der aktuellen Lage neben den gegenwärtigen Anforderungen auch bereits mehrere Schritte in die Zukunft gedacht werden, um solche Änderungen nachhaltig und wirtschaftlich zu gestalten. Lassen Sie uns gemeinsam einen Ausflug in den Dschungel der Anforderungen wagen.
Anforderungen der MDR (EU) 2017/745 an Werkstoffe und Stoffe
Der Hauptfokus der MDR liegt auf der Nachweiserbringung zur Sicherheit und Leistung eines Medizinprodukts über dessen gesamten Lebenszyklus. Welche Nachweise gefordert werden, lässt sich in den grundlegenden Sicherheits- und Leistungsanforderungen im Anhang I der MDR nachschlagen. In Kapitel II widmet sich der Abschnitt 10.4 komplett dem Thema „Stoffe“ und den Fragen, welche Stoffe hierbei erlaubt sind, sowie welche besondere Aufmerksamkeit bedürfen.
Generell sollen Medizinprodukte so ausgelegt und hergestellt werden, dass die Risiken durch Stoffe oder Partikel, die aus dem Produkt freigesetzt werden können, einschließlich Abrieb, Abbauprodukten und Verarbeitungsrückständen, so weit wie möglich verringert werden.
Wenn es sich um kritischere Medizinprodukte handelt, die
- invasiv angewandt werden und direkten Körperkontakt haben,
- dem Körper Arzneimittel, Körperflüssigkeiten oder sonstige Stoffe einschließlich Gase (wiederholt) verabreichen oder entnehmen oder
- solche Arzneimittel, Körperflüssigkeiten oder sonstige Stoffe einschließlich Gase, die dem Körper (wiederholt) verabreicht werden, transportieren oder lagern,
dann gelten zusätzliche Anforderungen in Bezug auf die Materialauswahl und den erlaubten Massenanteil, die im Abschnitt 10.4.1 und hier im Weiteren erläutert werden. Gesondert werden die Stoffe mit
- kanzerogenen (krebserregenden, carcinogenic),
- mutagenen (das Erbgut verändernden, mutagenic),
- reproduktionstoxischen (die Fruchtbarkeit bei Frau und Mann und/oder Entwicklung der Nachkommen beeinträchtigenden, toxic to reproduction),
- und endokrin wirkenden Eigenschaften (ED = endokrin disruptiv, endocrine disruptors), also mit Auswirkungen auf das Hormonsystem, erwähnt,
auch bekannt als CMR- oder ED-Stoffe. Haben vorgesehene Stoffe eine CMR-Klassifizierung der Kategorie 1A oder 1B („Kann Krebs erzeugen“/„Kann die Fruchtbarkeit beeinträchtigen“/„Kann vermutlich das Kind im Mutterleib schädigen“) oder endokrin disruptive Eigenschaften, dürfen sie nur in Konzentrationen < 0,1 Prozent Massenanteil verwendet werden. Sollen solche Stoffe in größeren Maßstäben verwendet werden und ist die direkte, invasive Anwendung vorgesehen, ist für den Einsatz eine Rechtfertigung notwendig, deren Inhalte in den Abschnitten 10.4.2 – 10.4.5 der MDR behandelt werden.
Generell muss bei einer solchen Rechtfertigung immer auf die neuesten wissenschaftlichen Erkenntnisse, weitere EU-Verordnungen oder Leitlinien eingegangen werden. Diese werden dann auf das eigene Medizinprodukt übertragen, um produktspezifisch eine fundierte Rechtfertigung und Bewertung zu erhalten. Sind kritische Stoffe in höherem Massenanteil in den Anwendungsteilen des Medizinprodukts enthalten, muss eine Kennzeichnung über das Vorhandensein solcher Stoffe auf den Produkten selbst oder der Einzelverpackung erfolgen. Eine genaue Auflistung der betroffenen Stoffe ist ebenfalls erforderlich. Patientengruppen, die als besonders anfällig für solche Stoffe und/oder Werkstoffe gelten, werden über Restrisiken und eventuelle Vorsichtsmaßnahmen in der Gebrauchsanweisung informiert.
Einfluss einer (Roh-)Materialänderung auf die biologische Sicherheit
Ist eine Änderung eines Rohmaterials für ein Medizinprodukt vorgesehen, bedeutet das nicht zwangsläufig, dass alle bisher gültigen Tests zum Nachweis der Biokompatibilität nicht mehr anwendbar sind. Vielmehr ist die Bewertung solcher Änderungen vorgelagert im Rahmen des Risikomanagementsystems gemäß EN ISO 14971 vorzunehmen. Dabei müssen die potenziellen Auswirkungen der Änderung betrachtet werden, die je nach vorgesehener Änderung unterschiedliche Ergebnisse liefern können. Werden hierbei nicht vertretbare Risiken identifiziert, sind die Hersteller dazu verpflichtet, diese im Rahmen der Anforderungen gemäß EN ISO 10993-1 in einer neuen Version des biologischen Bewertungsplans zu benennen. Zusätzlich muss eine wissenschaftliche Literaturrecherche in Bezug auf das vorgesehene neue Material und die vorgesehene klinische Anwendung des Medizinprodukts durchgeführt werden, da nur mit einer ausreichenden physikalischen und chemischen Charakterisierung eine fundierte Aussage über die biologische Sicherheit getroffen werden kann. Weiterhin müssen die Endpunkte, die sich ebenfalls aus der EN ISO 10993-1 – Annex A, Tabelle A.1 ergeben, betrachtet und in Bezug auf die vorgesehene Materialänderung bewertet werden. Sollte sich das Risiko im Anschluss immer noch im nicht akzeptablen Bereich befinden oder keine passenden Literaturdaten zu finden sein, dann folgt die Erstellung einer neuen Teststrategie mit der anschließenden finalen Bewertung im Rahmen der Erstellung einer neuen Version des biologischen Bewertungsberichts.
PFAS und nachhaltige Kunststoffe – aktuelle Möglichkeiten zum Einsatz
Neben der MDR und den dort genannten weiteren Anforderungen sollten auch immer neueste Entwicklungen und Beschlüsse innerhalb der EU betrachtet werden, da diese, auch wenn die MDR nicht direkt betroffen ist, durchaus eine Auswirkung auf Medizinprodukte haben können. Dies kann beispielsweise anhand der aktuellen Entwicklungen zur Nachhaltigkeit im Rahmen des EU Green Deal und der diskutierten Beschränkung von per- und polyfluorierten Alkylsubstanzen (PFAS) beobachtet werden.
Die Ziele des EU Green Deal benennen die Etablierung einer modernen, ressourceneffizienten und wettbewerbsfähigen Wirtschaft mit dem Ziel, bis 2050 keine Netto-Treibhausgase mehr zu emittieren. Auch hier müssen die Medizinprodukteindustrie und die jeweiligen Hersteller Auflagen einhalten. Hierbei besteht die Herausforderung, das technisch Mögliche regulatorisch sinnvoll und wirtschaftlich im Unternehmen zu integrieren.
Teil des Green Deal ist auch die Überarbeitung der REACH-Verordnung, bzw. generell eine neue Chemikalienstrategie. Diese zielt darauf ab, Mensch und Umwelt besser vor schädlichen Chemikalien zu schützen und Innovationen durch Förderung der Verwendung sichererer und nachhaltigerer Chemikalien voranzutreiben. Um dieses Ziel zu erreichen, wurde im Februar 2023 ein Vorschlag zur Beschränkung einer kompletten Stoffgruppe - der PFAS - bei der Europäischen Chemikalienagentur (ECHA) eingereicht.
PFAS weisen amphiphile Eigenschaften auf. Das heißt, sie haben neben hydrophoben Kohlenstoff-Fluor-Verbindungen auch hydrophile Anteile durch die Kopfgruppe. Aufgrund dieser amphiphilen Eigenschaften in Kombination mit der hohen Hitzebeständigkeit sind die Stoffe chemisch sehr inert (träge), reichern sich so in der Umwelt an und können weitreichende Auswirkungen auf die menschliche Gesundheit haben. Die Herstellung, Inverkehrbringung und Verwendung dieser Stoffgruppe soll nun durch den eingereichten Vorschlag sukzessive beschränkt werden. Dabei sieht der Verbotsvorschlag für Medizinprodukte eine Übergangsregelung von 13,5 Jahren vor (18 Monate plus 12 Jahre Ausnahmeregelung). Bis September 2023 konnten im Rahmen eines Konsultationsverfahrens Kommentare zu dem eingereichten Verbotsentwurf eingereicht werden, die von Fachausschüssen der ECHA gesichtet werden. Mit einer Stellungnahme der ECHA wird 2024 gerechnet, wobei sich bisher nur erahnen lässt, welche Änderungen eingebracht werden. Dennoch ist absehbar, dass die gesamte Stoffgruppe beschränkt und es lediglich für notwendige Bereiche, wie u. a. die Medizintechnikindustrie, Ausnahmen geben wird. Deshalb ist die Aufgabe der Medizinproduktehersteller nun klar – sich auf das bevorstehende Verbot bestmöglich vorzubereiten.
Unabhängig, ob es um den zukünftigen Einsatz der PFAS oder nachhaltigerer Kunststoffe wie biobasierten oder recyclierten Kunststoffen im Medizinprodukt geht: die gezielte Etablierung einer sinnvollen Datenlage ist immer vonnöten. Im Folgenden werden die verschiedenen Möglichkeiten aufgezeigt, wie zukünftig biobasierte bzw. recyclierte Kunststoffe in Medizinprodukten verwendet oder PFAS-haltige Medizinprodukte vorerst sicher auf den Markt gebracht werden können.
Per- und polyfluorierte Alkylsubstanzen – PFAS
Um der bevorstehenden Beschränkung der PFAS strategisch entgegen treten zu können, ist eine Auflistung des Produktportfolios inklusive der verwendeten Materialien unabdingbar. Werden hierbei essenzielle PFAS identifiziert, gilt es direkt zu handeln. Der erste Schritt ist immer die frühzeitige Kommunikation mit allen Stakeholdern, um sicherzustellen, dass z. B. Zulieferer auch weiterhin Rohstoffe liefern und nicht aufgrund der Beschränkung alle PFAS-Komponenten abkündigen. Der Ursprung der vollumfassenden PFAS-Beschränkung liegt neben den kritischen Eigenschaften auch in der häufig fehlenden Datenlage im Hinblick auf die Auswirkungen der PFAS auf Umwelt und Gesundheit, weshalb es wichtig sein wird, für Medizinprodukte eine durchdachte sowie maßgeschneiderte Bewertungsstrategie aufzusetzen, um alle potenziellen Risiken adäquat mindern zu können. Diese kann wie folgt aussehen:
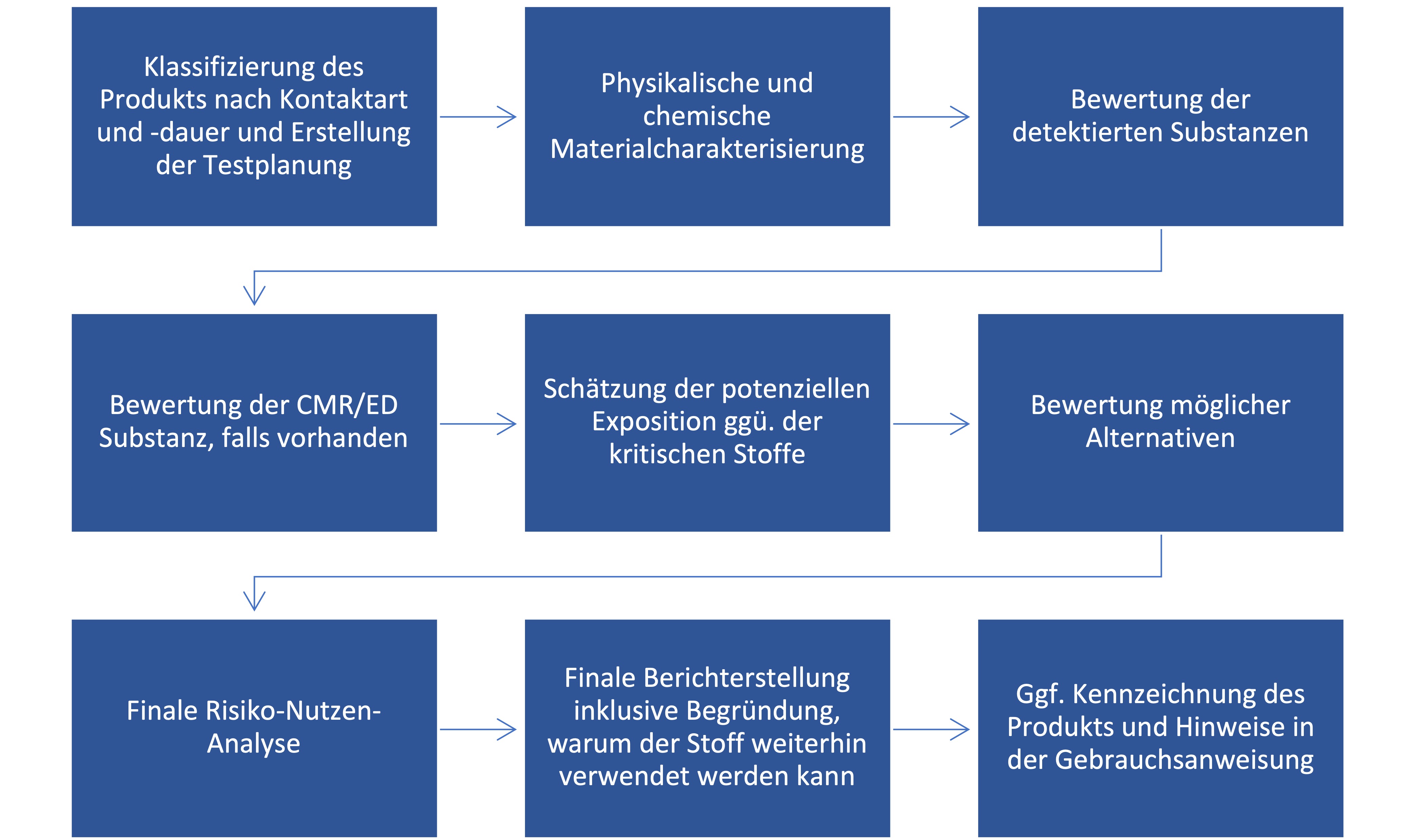 Mögliche Bewertungsstrategie für Medizinprodukte; Abkürzungen: CMR = Cancerogenic, Mutagenic, Toxic to Reproduction, ED = Endokrin Disruptiv; ggü. = gegenüber; ggf. = gegebenenfalls.
© Metecon GmbH
Mögliche Bewertungsstrategie für Medizinprodukte; Abkürzungen: CMR = Cancerogenic, Mutagenic, Toxic to Reproduction, ED = Endokrin Disruptiv; ggü. = gegenüber; ggf. = gegebenenfalls.
© Metecon GmbH
Die vorgestellte Strategie bietet nun die Möglichkeit, eine datenbasierte Bewertung von Medizinprodukten in Bezug auf deren klinischen Gebrauch durchzuführen. So kann ein Nachweis geliefert werden, dass der Gebrauch der Medizinprodukte trotz PFAS-Einsatz keine biologischen und toxikologischen Risiken birgt. Zusätzlich können in der Gebrauchsanweisung weitere Warn- und Entsorgungshinweise ergänzt werden, die die unkontrollierte Freisetzung einschränken, welche derzeit das Hauptproblem darstellt, und so ggf. eine Kreislaufwirtschaft oder zumindest eine kontrollierte Entsorgung der Medizinprodukte ermöglichen.
Durch diese Strategie kann vorerst sichergestellt werden, dass Sicherheit und Wirksamkeit bereits auf dem Markt befindlicher PFAS-haltiger Medizinprodukte gewährleistet sind. Langfristig ist es jedoch von entscheidender Bedeutung, dass der Einsatz von PFAS reduziert wird, und dass durch Forschung und Entwicklung Substitute für PFAS entwickelt werden, die in innovativen Medizinprodukten, aber auch bei bereits auf dem Markt befindlichen Produkten, berücksichtigt werden.
Darüber hinaus gibt es noch weitere Möglichkeiten für Hersteller, sich auf ein bevorstehendes PFAS-Verbot vorzubereiten:
- Modulierung der PFAS, um z. B. einen schnelleren toxikologisch unbedenklichen Abbau zu ermöglichen.
- Modulierung der Medizinprodukte, um eine potenzielle Freisetzung der PFAS einzuschränken.
- Hersteller und Lieferanten können eine medizinische Ausnahme der nötigen Stoffe beantragen (aufwendig).
- Registrierung und Zulassung der Stoffe bei der zuständigen Behörde als Interessenverband mehrerer Hersteller und Lieferanten/Zulieferer.
Die letzten beiden Punkte setzen jedoch eine genaue und aussagekräftige Charakterisierung des final eingesetzten Medizinprodukts und des verwendeten PFAS-Rohmaterials voraus.
Nachhaltigere Kunststoffe in Medizinprodukten
Ein Ziel des EU Green Deal ist die Reduktion der Netto-Treibhausgasemissionen bis zur kompletten Klimaneutralität im Jahr 2050. Aber auch die Abkopplung des Wachstums von der Ressourcennutzung und niemanden, weder Mensch noch Region, im Stich zu lassen, stehen im Fokus, um den Übergang zu einer modernen, ressourceneffizienten und wettbewerbsfähigen Wirtschaft zu schaffen. Dabei ist der EU Green Deal eng mit dem Begriff „Nachhaltigkeit“ verbunden. Die Dimensionen der Nachhaltigkeit umfassen neben der ökologischen auch die soziale und die wirtschaftliche Nachhaltigkeit. Diese beeinflussen sich gegenseitig und müssen gleichermaßen berücksichtigt werden.
Doch wie kann der vielumfassende Begriff „Nachhaltigkeit“ für die Medizinprodukteindustrie eingegrenzt werden? Ein denkbarer Ansatz, um den Inhalten des Green Deal Folge zu leisten, ist der Einsatz nachhaltiger Ressourcen, z. B.:
- Recycelte Kunststoffe: Diese bestehen aus wiederaufbereiteten Kunststoffabfällen, die meist in einem chemischen oder mechanischen Recyclingprozess gewonnen werden. Bei recycelten Kunststoffen wird zwischen Post-Consumer-Rezyklaten – recyceltem Kunststoff aus Haushaltsabfällen – und Post-Industrial-Rezyklaten – recyceltem Kunststoff aus „Abfällen“ anderer Industrien wie z. B. Speiseölraffinerien – unterschieden.
- Biobasierte Kunststoffe: Dabei handelt es sich um Kunststoffe aus nachwachsenden Rohstoffen.
- Biologisch abbaubare Kunststoffe: Zu den biologisch abbaubaren Kunststoffen zählen all jene Kunststoffe, die durch Mikroorganismen zersetzt werden können.
Die genannten Ansätze können allerdings aus regulatorischer Sicht schlagartig dezimiert werden, da beispielsweise beim Einsatz von Rezyklaten momentan noch keine gleichbleibende Qualität gewährleistet werden kann, die allerdings für den Nachweis der Sicherheits- und Leistungsanforderungen für Medizinprodukte notwendig ist. Hier ist eine Kosten-Nutzen-Bewertung sinnvoll. In Zukunft wäre der Einsatz von Rezyklaten und biobasierter Kunststoffe vermehrt möglich, da hier die Möglichkeit der Synthese von reinen Stoffen besteht. Trotzdem spricht die Praxis aktuell eine andere Sprache, da es auch hier einige Fallstricke zu beachten gibt:
- Die Verwendung von rein biobasierten Kunststoffen ist heute noch nicht gängig, aber erste Ansätze finden bereits ihren Weg in die Praxis. Dennoch werden aktuell meist Mischungen aus fossilen und biobasierten Kunststoffen in unterschiedlichen Anteilen verwendet. Grund für die Mischung ist, dass meist nur so die gewünschten Eigenschaften wie Stabilität, Reinheit sowie thermische und mechanische Belastbarkeit erreicht werden können. Es gibt aber auch Polymere wie Polyethylen, Polymilchsäuren (Polylactide), Polyhydroxyalkanoate (PAH), Polybutylensuccinat (PBS) oder Poly(-hydroxybutyrat-co-hydroxyvalerat)-Copolymere (PHBV), die zu 100 Prozent biobasiert eingesetzt werden können.
- Während die Verwendung von Kunststoffen mit Rezyklatanteil beispielsweise bereits bei Verpackungen von Medizinprodukten fallweise möglich ist, ist der Einsatz von Rezyklaten für höher klassifizierte Medizinprodukte ohne Standardisierung in der Herstellung von Rezyklaten z. B. durch gleichbleibende Qualität und immer gleichem Rezyklat-Anteil momentan nahezu unmöglich.
Diese beiden exemplarischen Ansätze verdeutlichen, dass der Einsatz biobasierter und recyclierter Kunststoffe nicht ausgeschlossen ist und in einigen Ansätzen bereits Anwendung findet. Allerdings wird auch deutlich, dass eine umfangreiche Charakterisierung und weitere Nachweise notwendig sind, um den Einsatz nachhaltiger Kunststoffe als biologisch sicher zu gewährleisten, und um die geforderten Sicherheits- und Leistungsanforderungen für Medizinprodukte zu erfüllen.
Anforderungen an die Nachweislage – was ist notwendig?
Allein diese beiden Beispiele verdeutlichen, dass die erforderliche Nachweislage sich in Zukunft nicht verringern wird. Auch im Hinblick auf die verschiedenen Regularien bleibt der Kampf durch einen Urwald aus Anforderungen nicht aus, der im Folgenden etwas übersichtlicher dargestellt wird:
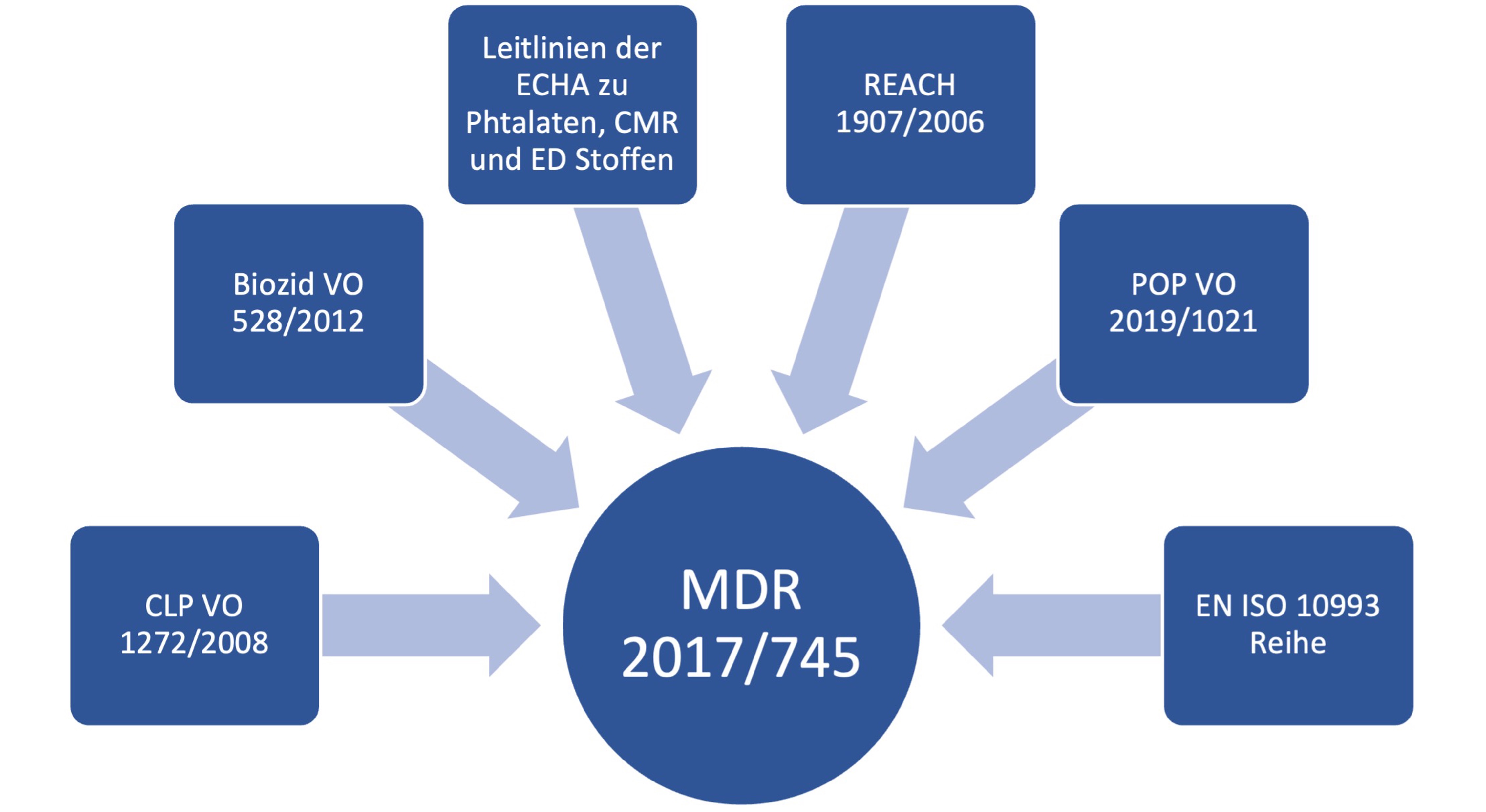 Übersicht weiterer Verordnungen, die zusätzlich gelten können. Abkürzungen: MDR = Medical Device Regulation; CLP VO = Verordnung zur Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP: Classification, Labeling, Packaging); ECHA = European Chemical Agency; REACH = Verordnung zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH: Registration, Evaluation, Authorisation and Restriction of Chemicals); POP VO = Verordnung über persistente organische Schadstoffe (POP = persistent organic pollutants); EN = Europäische Norm; ISO = International Organization for Standardization.
© Metecon GmbH
Übersicht weiterer Verordnungen, die zusätzlich gelten können. Abkürzungen: MDR = Medical Device Regulation; CLP VO = Verordnung zur Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP: Classification, Labeling, Packaging); ECHA = European Chemical Agency; REACH = Verordnung zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH: Registration, Evaluation, Authorisation and Restriction of Chemicals); POP VO = Verordnung über persistente organische Schadstoffe (POP = persistent organic pollutants); EN = Europäische Norm; ISO = International Organization for Standardization.
© Metecon GmbH
Im Falle der Medizinprodukte bleibt die MDR 2017/745 als Leitverordnung bestehen, weshalb sich an den Nachweisen zur biologischen Sicherheit gemäß den normativen Angaben der EN ISO 10993-Reihe primär nichts ändert. Dennoch wird auf weitere Verordnungen oder Veröffentlichungen wie der ECHA verwiesen. Diese Anforderungen gilt es zu berücksichtigen und sicherzustellen, dass die Lieferanten ihren Aufgaben im Rahmen von z. B. REACH nachkommen. Ebenfalls ergeben sich immer wieder Neuerungen und Ergänzungen der zusätzlichen Verordnungen, die Auswirkungen auf die Medizinprodukte mit sich bringen können, weshalb eine Auseinandersetzung mit den Verordnungen erfolgen muss.
Fazit
Die Zukunft der Medizinprodukte und die Anforderungen an die biologische Sicherheit werden sicherlich spannend bleiben, gerade im Hinblick auf potenzielle Verbote unter REACH, die vermutlich eher zu- als abnehmen werden. Nach Einführung der MDR im Jahr 2017 blieb zu Beginn der Aktionismus der Unternehmen aus, was zu Verzögerungen im Rahmen der Zertifizierungen geführt hat. Aus diesen Fehlern sollte nun gelernt werden, auch wenn es zunächst als großer, kostspieliger Aufwand erscheint. Deshalb sollten Hersteller nun ins Handeln kommen:
- Überblick über Rohstoffe verschaffen.
- Auflisten aller potenziell betroffener Rohstoffe/Medizinprodukte, welche von Änderungen durch Verbote nach REACH wie z. B. dem PFAS-Verbot betroffen wären, oder bei denen der Einsatz nachhaltiger Werkstoffe angestrebt wird.
- Priorisierung nach Wichtigkeit bzw. Notwendigkeit der Medizinprodukte. Wenn beispielsweise durch das Verbot von PFAS behandelbare Krankheiten nicht mehr behandelt werden können, dann haben solche Produkte Vorrang.
- Priorisierung gemäß Notwendigkeit des Einsatzes der verbotenen Stoffe/Materialien und Notwendigkeit der Änderung.
- Argumentationsgrundlage durch umfangreiche Risikoanalyse und Charakterisierung der vorgesehenen Stoffe und Medizinprodukte schaffen.
- „Verbündete“ suchen für Interessenverband und Beantragung medizinischer Ausnahmen.
- Nachhaltigkeit kann auf vielen Ebenen stattfinden und muss nicht immer direkt am kritischsten Punkt wie z. B. einer kompletten Materialänderung ansetzen – die Materialreduktion der fossilen Rohstoffe im Produkt selbst wäre ein denkbarer Ansatz.
- Biobasierte oder recyclierte Kunststoffe für den Einsatz in Medizinprodukten für Komponenten ohne direkten Körperkontakt als ersten Schritt nutzen, da sonst schwer ein sinnvolles Kosten-Nutzen-Verhältnis zu erreichen ist. Dieser Punkt sollte auch bei der Entwicklung neuer Produkte bereits von vorneherein mitgedacht werden.
Zusammenfassend ist eine vorgesehene Materialänderung, ob der Ursprung nun im Nachhaltigkeitsansatz oder in anstehenden Verboten von Stoffen liegt, durchaus möglich. Es bedarf lediglich einer sinnvollen Analyse der Situation, aus der dann eine von allen Seiten betrachtete Risikoanalyse hervorgeht. Nur so kann eine schlüssige Bewertung vorgenommen und bei Bedarf durch geeignete Prüfungen noch bestehende Risiken gemindert werden.