BIOPRO Spezial: Die Klinische Prüfung im Blick
Klinische Prüfung von Medizinprodukten: Welche Fehler es zu vermeiden gilt
Die steigende Notwendigkeit Klinischer Prüfungen durch Inkrafttreten der Verordnung (EU) 2017/745 im Jahr 2017 und der Anwendbarkeit ab Mai 2021 ist hinlänglich bekannt. Durch die strikteren Regulierungen müssen für mehr Medizinprodukte nun eigene klinische Daten erhoben werden. Dies betrifft nicht nur implantierbare Produkte und Medizinprodukte der Klasse III, sondern durch gestiegene Anforderungen an die Äquivalenzbewertung alle stofflichen Medizinprodukte und Software.
Die in Abschnitt "7. Unklarheit über regulatorische Anforderungen" dieses Beitrags zur Verfügung gestellten Informationen wurden am 26. Mai 2021 aktualisiert.
Die Verordnung (EU) 2017/745 über Medizinprodukte (Medical Device Regulation, MDR) definiert in Artikel 3 die Klinische Prüfung als „eine systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer einbezogen sind, und die zwecks Bewertung der Sicherheit oder Leistung eines Produkts durchgeführt wird“.1
Um diese systematische Untersuchung regulatorisch korrekt durchzuführen, werden die Hersteller betroffener Medizinprodukte durch die MDR und die ISO 14155, die 2020 im Hinblick auf die MDR als neue Ausgabe veröffentlicht wurde, mit zahlreichen zusätzlichen, und zum Teil auch neuen Aufgaben konfrontiert, die in vielen Fällen zu einer großen Herausforderung führen.2
Neben der Unklarheit über die Notwendigkeit einer Klinischen Prüfung und fehlenden Kenntnissen bezüglich der Planung und Umsetzung eines solchen Vorhabens, mangelt es häufig an Wissen bezüglich der regulatorischen Anforderungen. Dies bedeutet für die Hersteller meist nicht nur einen höheren zeitlichen Aufwand, sondern auch höhere Kosten.
1. Unklarheit über die Notwendigkeit einer Klinischen Prüfung
Seit Inkrafttreten der MDR wurde versucht, das Bewusstsein für die Bedeutung klinischer Daten durch die MDR zu schärfen, doch wissen viele Hersteller immer noch nicht, ob eine Klinische Prüfung überhaupt notwendig ist.
In Artikel 61, in Verbindung mit Anhang XIV, fordert die MDR einen Plan für die Klinische Bewertung. In diesem Plan soll sich der Hersteller im Rahmen eines klinischen Entwicklungsplans mit klinischen Daten und deren Erhebung im Hinblick auf das Medizinprodukt auseinandersetzen. Der Plan für die Klinische Bewertung wird bereits zu Beginn der Entwicklung erstellt und legt die Basis für den kontinuierlichen Prozess der Datenerhebung für das Medizinprodukt.
Somit soll sichergestellt werden, dass eine ggf. erforderliche Klinische Prüfung schon frühzeitig im Entwicklungsprozess eingeplant wird. Denn nichts ist fataler als im Rahmen der Erstellung des Berichts zur Klinischen Bewertung festzustellen, dass die Datenlage nicht ausreicht, um Nutzen, Leistung und Sicherheit des Medizinprodukts eingehend zu belegen.
Auch im Rahmen der Klinischen Nachbeobachtung (Post Market Clinical Follow-up, PMCF) kann eine Erhebung eigener klinischer Daten zum Medizinprodukt erforderlich werden. Beispielsweise, wenn der Indikationsbereich oder die Zweckbestimmung erweitert werden soll – dies sind signifikante Änderungen des Produkts mit Auswirkung auf Nutzen, Leistung und Sicherheit, die ebenfalls mit klinischen Daten zu belegen sind. Fehlen klinische Daten hierzu, wird auch dann eine Klinische Prüfung nach Artikel 74 der MDR erforderlich. Andernfalls dürfen die Änderungen nicht vorgenommen werden.
2. Unpräzise oder falsche Ziele
Eine sorgfältige Planung der Klinischen Prüfung ist essenziell, damit am Ende das gewünschte Ergebnis herauskommt: nämlich klinische Daten, die die klinische Leistung, Sicherheit oder den klinischen Nutzen des jeweiligen Medizinproduktes belegen. So kann das Ziel beispielsweise der Nachweis der klinischen Leistung (Wirksamkeit) des Medizinprodukts bei der Linderung von Schmerzen sein. Das Ergebnis ist dann die mit klinischen Daten belegte Wirksamkeit – somit liefert der Hersteller einen Beleg für die Behauptung, dass das Medizinprodukt zur Schmerzlinderung bei einer bestimmten Indikation beiträgt, wenn es gemäß seiner Zweckbestimmung angewendet wird.
Zu Beginn jeder Klinischen Prüfung muss deshalb zunächst das Ziel, das sogenannte „Objective“, festgelegt werden. Daraus leiten sich dann die zu evaluierenden Parameter zu Leistung, Sicherheit und zum Nutzen des Produkts ab. Diese sind im Rahmen der Klinischen Prüfung die Endpunkte, die sich ihrerseits in primäre und sekundäre Endpunkte aufteilen.
Endpunkt
Der primäre Endpunkt ist das Kernstück der Klinischen Prüfung zum Erreichen des Ziels. Auf ihm basiert die Fallzahlplanung, die zum Beispiel Anhaltspunkte für die Populationsgröße und die Anzahl der erforderlichen Prüfzentren gibt.
Sekundäre Endpunkte unterstützen den primären Endpunkt und liefern weitere wichtige Informationen und klinische Daten für zusätzliche Aspekte. So kann beispielsweise die Sicherheit des Medizinprodukts (Anzahl/Häufigkeit von Nebenwirkungen, Komplikationen, schwerwiegenden unerwünschten Ereignissen) weitere Hinweise unter anderem auch für das Risikomanagement liefern.
Die Hersteller müssen genau festlegen, welche Endpunkte die Studie mit klinischen Daten belegen muss. Andernfalls besteht die Gefahr, dass die Studie zwar eine Hypothese beweist, dieser Beweis aber nicht geeignet ist, um die Sicherheit, die Leistungsfähigkeit und den klinischen Nutzen des Medizinprodukts zu belegen.
Ziele einer Klinischen Prüfung nach Artikel 62 der MDR (Klinische Prüfungen, die zum Nachweis der Konformität des Produktes durchgeführt werden, also Zulassungsstudien) können somit beispielsweise folgende sein:
- Bewertung der Leistung (Wirksamkeit) und Sicherheit des Produkts bei der Schmerzlinderung,
- Bewertung der langfristigen Leistung (Wirksamkeit) und Sicherheit eines Implantats und dessen Bruchfestigkeit,
- Bewertung der Leistung (Wirksamkeit) und Sicherheit des Produkts bei der Behandlung von einer in der Zweckbestimmung genannten Behandlung.
Bei Klinischen Prüfungen von Produkten, die bereits eine CE-Kennzeichnung tragen (Artikel 74 der MDR, PMCF-Studien), sollen meist neue Daten erhoben oder Datenlücken geschlossen werden, die die Leistung (Wirksamkeit) und Sicherheit des zu evaluierenden Produkts bestätigen.
Das Ziel wird zunächst also eher generalisiert formuliert; die Endpunkte hingegen leiten sich konkreter ab, wie die folgenden Beispiele zeigen:
Primärer Endpunkt
- ist bei Patienten mit Knorpelschäden im Knie der IKDC-Score (International Knee Documentation Comitee, IKDC) und eine Verbesserung über 6 und 12 Monate,
- ist die positive Zunahme der Lebensqualität versus Placebo,
- ist die größere Schmerzlinderung bei Anwendung des Produkts und Standardtherapie versus Standardtherapie alleine.
3. Falsches Studiendesign
In die Überlegungen zu den Endpunkten, die das Ziel der Klinischen Prüfung näher definieren, fließen Überlegungen zum Design der Studie ein.
Das Studiendesign kann prospektiv oder retrospektiv aufgebaut werden:
- Prospektiv (in die Zukunft gerichtet): vergleichend oder nicht vergleichend (mit oder ohne Kontrollgruppe) – wenn die Studie mit einer Kontrollgruppe durchgeführt wird, kann eine Zuteilung randomisiert oder nicht randomisiert erfolgen. In diesem Kontext muss auch festgelegt werden, ob eine Nichtunterlegenheit („Non-inferiority“), Überlegenheit („Superiority“) oder eine Äquivalenz im Falle einer Kontrollgruppe gezeigt werden soll. Unter einer Nichtunterlegenheit versteht man dabei, dass das Prüfprodukt nicht wesentlich schlechter als das Kontrollprodukt ist. Eine Überlegenheit zeigt das bessere Abschneiden des Prüfprodukts versus der Kontrolle. Die Äquivalenz sagt aus, dass das Prüfprodukt im Wesentlichen „gleich gut“ wie das Kontrollprodukt oder die Kontrollanwendung ist.
- Retrospektiv (Auswertung bereits vorhandener Daten): Hier ist vorausgesetzt, dass bereits Daten zum Produkt vorhanden sind, es also in der Anwendung ist. Dies trifft für neue Zulassungen nicht zu und findet deshalb eher im PMCF-Bereich Anwendung.
Die folgende Tabelle listet weitere Studiendesigns auf:
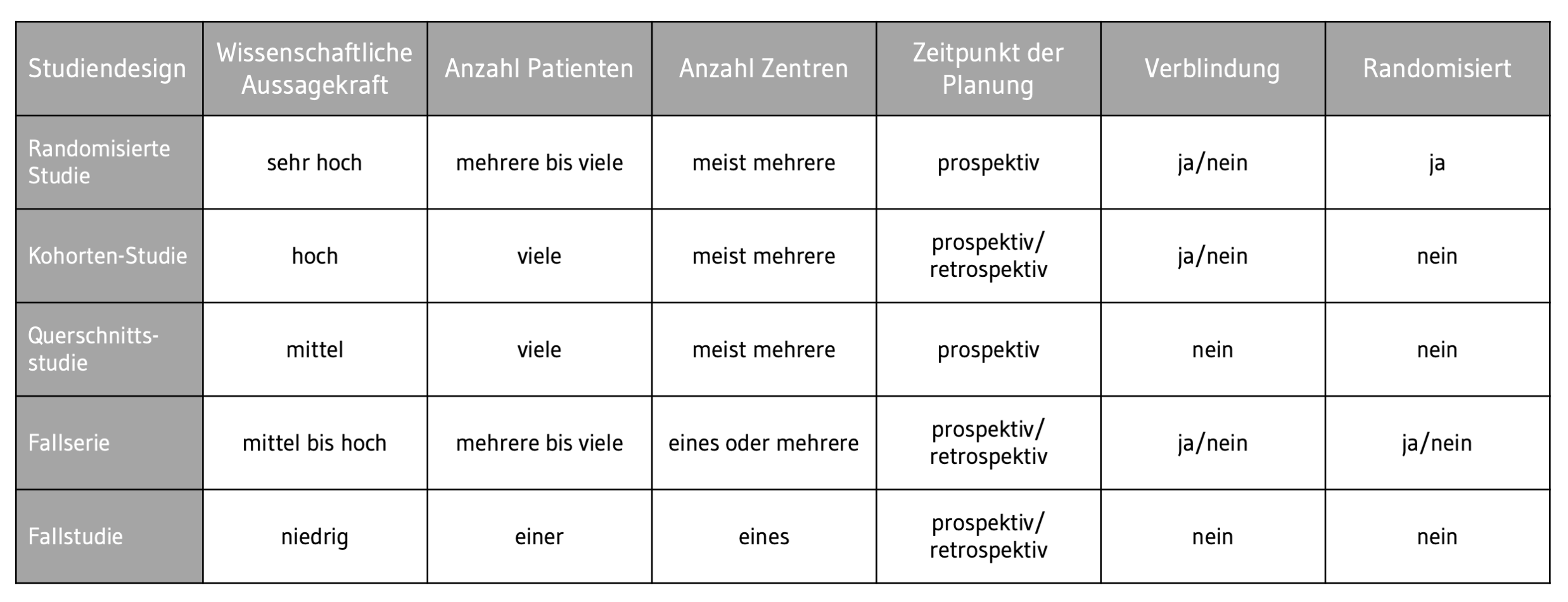 Tab. 1 Mögliche Designs einer Klinischen Prüfung. © Daniela Penn
Tab. 1 Mögliche Designs einer Klinischen Prüfung. © Daniela PennFehlendes Wissen, wie man mit dem Werkzeug „Design“ entsprechende Ziele erreicht, kann leicht dazu führen, dass hier der falsche Weg eingeschlagen wird und beispielsweise eine randomisierte kontrollierte Studie (Randomized Controlled Trial, RCT) durchgeführt wird, die mehr kostet und länger dauert, obwohl dasselbe Ergebnis auch mit einer prospektiven Fallserie erreicht worden wäre. Derzeit wird sich häufig für eine RCT entschieden, da dies als „Goldstandard“ eingeordnet wird. Dies hängt jedoch immer vom Medizinprodukt und der entsprechenden Fragestellung ab, die man beantworten möchte – und somit auch von den klinischen Daten, die erhoben werden sollen.
4. Keine ausreichende Teilnehmerzahl
Die statistische Fallzahlplanung ist nach Festlegung von Ziel, Endpunkten und Studiendesign das letzte wichtige Glied einer regulatorisch korrekten Planung der Klinischen Prüfung.
Basierend auf dem primären Endpunkt wird festgelegt, wie viele Probanden eingeschlossen werden müssen, um einen relevanten Effekt nachzuweisen – somit entscheidet sich darauf basierend letztendlich auch das Gelingen oder Scheitern einer Studie.
Für den Nachweis der Wirksamkeit einer Klinischen Prüfung, sowohl bei PMCF- als auch bei Zulassungsstudien, werden Hypothesen anhand des primären Endpunkts überprüft. Eine nachzuweisende Hypothese (Alternativhypothese) kann zum Beispiel die Überlegenheit eines Produkts („Superiority“) gegenüber einer Standardtherapie sein. Die Bestätigung oder Verwerfung einer Hypothese wird anhand erhobener Daten beurteilt und auf die Ergebnisse für die Grundgesamtheit, also auf die gesamte Zielgruppe, übertragen. Damit das Ergebnis aussagekräftig ist, müssen genügend Daten von Beobachtungen aus der Zielgruppe vorliegen. Liegen zu wenige Beobachtungen und somit klinische Daten vor, können tatsächlich vorhandene Behandlungseffekte nicht nachgewiesen werden, die Studie scheitert. Auf der anderen Seite führt ein großer Stichprobenumfang zu hohen Kosten, ist ethisch nur schwer begründbar, bindet Ressourcen und verlängert die Studiendauer.
Die Notwendigkeit einer Fallzahlplanung in der Planungsphase Klinischer Prüfungen ist zudem gesetzlich vorgeschrieben und wird durch die zuständige Ethikkommission überprüft. Die Berechnung des Stichprobenumfangs ist ein essenzieller Teil des klinischen Prüfplans (Clinical Investigation Plan, CIP) sowie des statistischen Analyseplans, einem wesentlichen Dokument für die spätere Auswertung der Daten. Hier sollte deshalb ein Biometriker unterstützen.
Für die Auswahl des geeigneten statistischen Tests ist die Art der Fragestellung wesentlich. Je nachdem, ob Überlegenheit oder Äquivalenz einer Behandlung nachgewiesen werden soll, sind andere Testverfahren erforderlich. Auch das Skalenniveau der primären Zielvariablen spielt eine entscheidende Rolle. Für nominale Merkmale (ja/nein, Erfolg/kein Erfolg) werden andere Verfahren eingesetzt als für ordinale (z. B. Likert-Skala) oder stetige Merkmale (z. B. visuelle Analogskala (VAS), Summenscores, etc.).
Effektgröße
Mit der Effektgröße wird der nachzuweisende, relevante Unterschied angegeben. Je nach Testverfahren werden verschiedene Maße verwendet. Als bekannteste Effektgröße gilt bei stetigen Variablen Cohens d, das den Unterschied zweier unabhängiger Gruppen in Relation zur gemeinsamen Streuung angibt.
Für kategoriale Endpunkte wird die Effektgröße W verwendet, die sich als Wurzel der quadrierten relativen Differenz der Proportionen ergibt.
Nach Cohen (1988)3 gelten dabei grob folgende Faustregeln:
Effektstärke ≈ 0,2: kleiner Effekt
Effektstärke ≈ 0,5: mittlerer Effekt
Effektstärke ≈ 0,8: großer Effekt
Für die Festlegung der Effektgröße werden möglichst präzise Vorinformationen aus der Literatur bzw. eigenen Pilotstudien benötigt. Ebenso fließt der medizinisch und praktisch relevante, nachzuweisende Unterschied ein. Eine mittlere Blutdrucksenkung von wenigen mmHg, also eine sehr kleine Effektstärke, kann zwar mit genügend hohem Stichprobenumfang statistisch nachgewiesen werden, ist aber praktisch für Patient und Mediziner irrelevant.
Signifikanzniveau des statistischen Tests
Das Signifikanzniveau a muss vorab festgelegt und im Studienprotokoll und im Statistischen Analyseplan (SAP) niedergeschrieben werden. Das Signifikanzniveau gibt die Wahrscheinlichkeit an, mit der man ein statistisch signifikantes Testergebnis erhält, sofern in der Zielgruppe tatsächlich kein Effekt vorhanden ist. Weiter wird unterschieden, ob der Test ein- oder zweiseitig durchgeführt wird. Einseitige Tests überprüfen Überlegenheitshypothesen. Üblich sind zweiseitige Fragestellungen, die hinsichtlich eines Unterschieds zweier Therapien einen Vergleich durchführen. Als Signifikanzniveau hat sich der Wert a = 5 % etabliert, bei einseitiger Fragestellung wird oft a = 2,5 % verwendet.
Power oder Macht
In der Planungsphase wird auch die Power oder Macht der Studie festgelegt. Darunter versteht man die Wahrscheinlichkeit, dass ein statistischer Test den tatsächlich vorhandenen Unterschied nachweist, also einen signifikanten p-Wert liefert. Die Macht einer Studie sollte also möglichst hoch sein. Hier sind Werte zwischen 80 % und 90 % üblich. Je höher die Power einer Studie, desto höher ist die resultierende Fallzahl.
Ziel- und Endpunktdefinition, Festlegung des Studiendesigns und Fallzahlplanung gehören somit zur strukturierten und umfassenden Planung der Klinischen Prüfung. Das falsche Ziel, falsche Endpunkte oder ein nicht passendes Design können zum Scheitern einer Klinischen Prüfung beitragen. Auch ein agiles, iterativ inkrementelles Vorgehen ist im Kontext Klinischer Prüfungen nicht zielführend. Nur auf einer sorgfältigen und umfassenden Planung kann ein durchdachtes, zielführendes Studienkonzept aufgebaut werden.
Nicht nur bei der Planung der Klinischen Prüfung, auch bei der eigentlichen Durchführung, der praktischen Umsetzung des klinischen Prüfplans, wird ein Hersteller, der im Rahmen der Klinischen Prüfung in der Regel die Rolle des Sponsors übernimmt, mit mehreren Herausforderungen konfrontiert.
5. Unzureichendes Monitoring und Datenmanagement
Fehler und Lücken bei der Datenerhebung und eine Datenerfassung, die nicht dem klinischen Prüfplan entspricht, machen die Aussagekraft einer Klinischen Prüfung zunichte. Ein engmaschiges Monitoring und ein fundiertes Datenmanagement sind für die Klinische Prüfung daher unerlässlich.
Ein korrektes Datenmanagement und das Monitoring der Klinischen Prüfung, also die Überwachung der korrekten Durchführung und prüfplangerechten Umsetzung der Klinischen Prüfung vor Ort im Prüfzentrum, sind regulatorisch gefordert. Nicht nur in der MDR, sondern auch in der ISO 14155 und damit auch gleichzeitig durch die Good Clinical Practice (GCP).
Beides, Datenmanagement und Monitoring, ist ebenfalls sorgfältig in einem Datenmanagement- und Monitoringplan auszuarbeiten und dann gemäß dem Plan entsprechend umzusetzen.
ISO 14155:2020-072
Kapitel A8 der ISO 14155:2020-07 umfasst das Datenmanagement:
„a) Verfahren Dateneingabe und Datensammlung (zum Beispiel CRF).
b) Verfahren zur Datenüberprüfung, Säuberung von Datenbanken und zum Starten und Ausführen von Datenabfragen. Insbesondere sind zeitnahe und zuverlässige Prozesse für die Erfassung von Daten und die Korrektur von Fehlern und Auslassungen erforderlich, um die Erstellung einer Datenbank von hoher Qualität und die Erreichung der Zielstellungen der Klinischen Prüfung durch die Umsetzung der geplanten Analyse sicherzustellen.
c) Gegebenenfalls Verfahren zur Verifizierung, Validierung und Sicherung elektronischer Datensysteme.
d) Verfahren zur Wahrung der Vertraulichkeit und zum Schutz personenbezogener Daten.
e) Verfahren zur Datenbanksperrung zu Beginn der Analyse und Speicherung nach Abschluss der Klinischen Prüfung.
f) Verfahren zur Datenspeicherung.
g) Festgelegte Aufbewahrungszeit.
h) Gegebenenfalls sonstige Aspekte der Qualitätssicherung“.
Die ISO 14155:2020-07 detailliert das Monitoring in Kapitel 9.2.4:
„Das Monitoring muss in Übereinstimmung mit dem Monitoringplan erfolgen. Der Zweck des Monitorings einer Klinischen Prüfung ist es, zu verifizieren, dass
a) die Rechte und das Wohlbefinden der Versuchspersonen geschützt werden,
b) die angegebenen Daten genau, vollständig und anhand der Quelldokumente verifizierbar sind und
c) die Durchführung der Klinischen Prüfung in Übereinstimmung mit dem bestätigten CIP, späteren Änderungen, der vorliegenden Internationalen Norm und den anwendbaren Anforderungen der EC [Ethikkommission] erfolgt“.
6. Falsches Prüfprodukt
Die Ergebnisse einer Klinischen Prüfung beziehen sich immer auf das Medizinprodukt, mit dem die Klinische Prüfung durchgeführt wurde. Gerade bei Zulassungsstudien muss die Klinische Prüfung mit dem finalen Produkt und nicht etwa mit einem Prototyp durchgeführt werden.
Klinische Daten zu einem Prototyp sind nicht als äquivalent zu denen des fertig entwickelten, finalen Medizinprodukts anzusehen und haben keinerlei Aussagekraft zur klinischen Leistung, Sicherheit oder zum Nutzen des finalen Produkts.
7. Unklarheit über regulatorische Anforderungen
Die MDR und das Medizinprodukterecht-Durchführungsgesetz (MPDG) haben die Anforderungen an „Klinische Prüfungen“ (MDR, Artikel 62 und 74) und „Sonstige Klinische Prüfungen“ eindeutig definiert und festgelegt.1,4
Bisher reguliert der 4. Abschnitt des Medizinproduktegesetzes (MPG) die Klinische Bewertung, Klinische Prüfung von Medizinprodukten sowie die Leistungsbewertungsprüfung mit allgemeinen und besonderen Voraussetzungen (MPG, §§20 und 21), Genehmigungsverfahren bei der Behörde und Ethikkommission (MPG, §22) sowie die Durchführung (MPG, §23).5 Diese §§20 bis 23b des MPG werden mit der Anwendbarkeit der MDR und damit auch des MPDG abgelöst. Artikel 62ff der MDR regulieren nun die Durchführung Klinischer Prüfungen von Medizinprodukten zur Erlangung einer CE-Kennzeichnung. Unklar war lange, wie die MDR bezüglich Klinischer Prüfungen mit CE-gekennzeichneten Produkten innerhalb ihrer Zweckbestimmung und ohne zusätzliche belastende oder invasive Untersuchungen interpretiert werden muss. In einer virtuellen Veranstaltung informierte das BfArM gemeinsam mit dem Bundesministerium für Gesundheit und dem Arbeitskreis der Ethik-Kommissionen am 5. Mai 2021, dass für Klinische Prüfungen mit diesen Produkten eine berufsrechtliche Beratung nach §15 BO (Berufsordnung der Ärzte, MBO-Ä6) ausreicht. Für Klinische Prüfungen mit CE-gekennzeichneten Produkten, die außerhalb der Zweckbestimmung durchgeführt werden oder bei denen zusätzliche belastende oder invasive Untersuchungen durchgeführt werden, ist Artikel 74 der MDR anzuwenden, der ein Ethikvotum vorschreibt. Artikel 74 der MDR gliedert PMCF-Studien innerhalb der Zweckbestimmung aus und betrachtet diese weiterhin wie schon im MPG als Ausnahme zu den Klinischen Prüfungen.
DiGA-Studien
Gerade bei Software spielt seit 2020 eine weitere Anforderung eine Rolle, wenn diese in Deutschland als digitale Gesundheitsanwendung (DiGA) gelistet und damit erstattungsfähig werden soll – in diesem Fall wird nämlich auch für Software der Klasse I und IIa eine sogenannte DiGA-Studie zum Nachweis des positiven Versorgungseffektes gefordert.
Eine DiGA-Studie ist eine nationale Besonderheit, schon alleine deshalb, weil sie nur in Deutschland durchgeführt werden kann. Zudem handelt es sich dabei meist um eine Studie an Medizinprodukten, für die normalerweise, bzw. in der Regel im Rahmen der Erfüllung der grundlegenden Sicherheits- und Leistungsanforderungen, und bei deren Nachweis in der Klinischen Bewertung für Medizinprodukte auf klinische Daten verzichtet werden kann. Stattdessen werden Leistungsdaten herangezogen.
Aufgrund dieser neuen Anforderung an diese Medizinprodukte ist zu überlegen, ob für diesen Zweck erhobene klinische Daten auch im Rahmen der MDR-Anforderungen an klinische Daten zum Medizinprodukt Anwendung finden können. Bei einer DiGA sollten die regulatorischen Anforderungen an das Medizinprodukt mit den Anforderungen an die DiGA im Hinblick auf die Klinische Prüfung (DiGA-Studie) miteinander verbunden werden, da die Klinische Prüfung für beide Bereiche genutzt werden kann.
Fazit
Zukünftig werden viele Hersteller mit dem Thema „Klinische Prüfung“ konfrontiert werden. Damit diese auch den erhofften Erfolg bringt und das angestrebte Ziel wie beispielsweise die Zulassung nach MDR entsprechend erreicht wird, sollte eine Klinische Prüfung sorgfältig geplant und regulatorisch korrekt durchgeführt werden.
Wir machen ausdrücklich darauf aufmerksam, dass unser Webangebot lediglich dem unverbindlichen Informationszweck dient. Alle angebotenen Informationen sind ohne Gewähr und erheben keinen Anspruch auf Vollständigkeit und Richtigkeit. Eine Haftung für die Angaben sowie für deren Rechtsverbindlichkeit wird nicht übernommen.