Klinische Prüfungen bei Humanarzneimitteln: Rechtsrahmen und Pläne der Politik
Klinische Prüfungen sind unerlässlich, um die Sicherheit und Wirksamkeit von Arzneimitteln zu gewährleisten. Doch welche rechtlichen Voraussetzungen gelten für ihre Genehmigung und Durchführung? Und welche Neuerungen plant die Bundesregierung?
Spätestens seit der Corona-Pandemie ist klar, wie wichtig die Forschung an neuen Wirkstoffen ist. Aber auch jenseits von COVID-19 werden neue Medikamente regelmäßig dringend gebraucht: etwa wegen der steigenden Zahl an antibiotikaresistenten Keimen und Zivilisationskrankheiten oder schlicht zur besseren Versorgung von alltäglichen Leiden wie einer Erkältung. Doch bevor ein neues Arzneimittel auf den Markt kommt, muss sichergestellt sein, dass es nicht nur zuverlässig wirkt, sondern auch sicher ist. Genau dies wird in Klinischen Prüfungen kontrolliert. Hierbei handelt es sich um Klinische Studien am Menschen, mit Medikamenten, die in der Entwicklung bereits weiter fortgeschritten sind und sich unter anderem in präklinischen Tests bereits bewährt haben. Solche Prüfungen sind Voraussetzung für die Zulassung von Medikamenten.
Rechtlicher Rahmen für Klinische Prüfungen in der EU
Dabei gelten jedoch strenge rechtliche und ethische Regeln. In Europa gibt die Verordnung über Klinische Prüfungen mit Humanarzneimitteln (EU) Nr. 536/20141) (auch Clinical Trials Regulation, CTR) den Rahmen vor. Bereits im Jahr 2014 erlassen, ist sie nun seit 31. Januar 2022 verbindlich anzuwenden. Als EU-Verordnung steht sie dabei auch über dem deutschen Recht. Klinische Prüfungen, die vor dem 31. Januar 2022 nach altem Recht begonnen wurden, dürfen allerdings noch bis 31. Januar 2025 nach altem Recht beendet werden. Danach gilt auch für sie die Clinical Trials Regulation.
Neben der CTR müssen Sponsoren außerdem nationale Regelungen, die die EU-Vorschriften teils ergänzen oder ausgestalten (in Deutschland etwa das AMG) sowie verschiedene Leitlinien und weitere ergänzende Dokumente beachten:
Clinical Trials Regulation
 Klinische Studien sind für sichere und wirksame Medikamente und damit eine bessere Versorgung von Patienten unerlässlich. © Ed Us/ Unspalsh
Klinische Studien sind für sichere und wirksame Medikamente und damit eine bessere Versorgung von Patienten unerlässlich. © Ed Us/ UnspalshDie Clinical Trials Regulation ist das maßgebliche rechtliche Instrument, nach dem sich Klinische Prüfungen in Europa richten. Auch die entsprechenden Vorschriften im deutschen Arzneimittelgesetz (§§ 40‐42c AMG) verweisen auf die CTR, ergänzen deren Regelungen jedoch teilweise.
Wichtig zu wissen: Die CTR gilt nicht für alle Klinischen Studien am Menschen, denn sie unterscheidet zwischen „Klinische Prüfungen“ und anderen Klinischen Studien. Klinische Prüfungen sind Studien am Menschen laut CTR nur, wenn gezielt eine neue Behandlung getestet wird und diese über die normale klinische Praxis hinausgeht. Alle anderen Klinischen Studien, also insbesondere nichtinterventionelle Studien, wie etwa Anwendungsbeobachtungen6), fallen daher nicht unter die Clinical Trials Regulation.
Allgemeine Grundsätze der CTR
Als allgemeinen Grundsatz stellt die CTR die Sicherheit, das Wohlergehen und die Rechte der Probandinnen und Probanden bei Klinischen Prüfungen in den Mittelpunkt. Daher sind Pflichten zur Aufklärung der Patientinnen und Patienten und ethische Prinzipien bei der Durchführung wie u.a. die Berücksichtigung der internationalen Standards der „Guten Klinischen Praxis“ (Good Clinical Practice, GCP)4) zentrale Bestandteile der Verordnung. Für die Genehmigung ist aus diesem Grund auch nicht nur das Okay der zuständigen Behörde, in Deutschland in der Regel das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) bzw. das Paul-Ehrlich-Institut (PEI), nötig, sondern auch das einer Ethikkommission. Die Klinische Prüfung muss außerdem immer so konzipiert sein, dass sie zuverlässige und belastbare Daten liefert.
Einreichungs- und Genehmigungsprozess der CTR
Eines der Ziele der CTR war, die Vorgaben für Klinische Prüfungen EU-weit anzugleichen, multinationale Studien zu erleichtern und dabei bürokratische Hürden möglichst niedrig zu halten. Dies hat sich auch auf den Genehmigungsprozess ausgewirkt.
Einreichen über CTIS
Eine zentrale Neuerung der Verordnung ist die Digitalisierung des Antragsverfahrens durch das Portal Clinical Trials Information System (CTIS)7), über das Sponsoren seit 31. Januar 2023 ihre Anträge für alle europäischen Länder zentral einreichen müssen, und zwar auch dann, wenn sie die Prüfung nur in einem EU-Land durchführen möchten. Dabei ist auch für multinationale Prüfungen nur ein Antrag nötig. Sonstige Kommunikation von Formalien wie Bescheide und Reportings erfolgen ebenfalls über CTIS.
Die EMA hat eine Reihe von Schulungen und Vorlagen8) für das Antragsverfahren und die CTIS-Plattform zur Verfügung gestellt, die die wichtigsten Prozesse wie Antragseinreichung oder Reporting Schritt-für-Schritt erklären.
Unterlagen Clinical Trial Application (CTA):
Für Anträge in Bezug auf klinische Prüfungen müssen Sponsoren über CTIS die folgenden Unterlagen einreichen bzw. die folgenden Angaben machen:
Form: In diesen Abschnitt gehören allgemeine Informationen zur klinischen Prüfung, z. B. das Anschreiben, Zahlungsnachweise, Nachweise über die Einhaltung von Datenschutzbestimmungen sowie die Veröffentlichungsdaten der Studien-Informationen.
Member State Concerned (betroffener Mitgliedsstaat, cMS): Sponsoren müssen angeben, in welchem Mitgliedsstaat oder welchen -Staaten sie die Prüfung durchführen möchten, und wie viele Probandinnen und Probanden dafür jeweils rekrutiert werden sollen.
Bei multinationalen Studien müssen Sponsoren dabei einen Reporting Member State (berichterstattenden Mitgliedsstaat, rMS) vorschlagen. Dieser spielt die führende Rolle während der Laufzeit der Klinischen Prüfung, leitet das Verfahren und gibt die Schlussfolgerung zu Part I des Bewertungsberichts heraus. Die finale Auswahl des rMS treffen die EU-Staaten untereinander.
Bewertungsbericht Part I: Der Genehmigungsantrag umfasst einen Bewertungsbericht Part I (Art. 6 CTR) und Part II (Art. 7 CTR). Part I befasst sich vor allem mit allgemeinen wissenschaftlichen Aspekten der Prüfung, die für alle Mitgliedsstaaten gelten. Hierzu zählt etwa das Studiendesign, Angaben zum therapeutischen Nutzen, die Prüferbroschüre etc. Part I lässt sich zunächst auch separat einreichen. Part II muss dann spätestens zwei Jahre später vorliegen.
Bewertungsbericht Part II: Dieser befasst sich mit Aspekten, die für die einzelnen Mitgliedsstaaten besonders relevant sind, in denen die Prüfung durchgeführt wird. Hier geht es um Einwilligung, Aufklärung, Rekrutierung sowie Entschädigung von Prüfungsteilnehmenden, Datenschutz, Eignung der Probandinnen und Probanden, Eignung der Prüfstellen und die Handhabung biologischer Proben
Details zu den erforderlichen Antragsunterlagen finden sich in Anhang I der CTR. Eine kompakte Übersicht über die in CTIS einzureichenden Unterlagen9) liefert die EMA in ihren Online-Schulungen.
Prüfung:
Die Koordination der Genehmigung übernimmt die zuständige Behörde im jeweiligen EU-Staat. Für die Entscheidung haben die Behörden dann insgesamt maximal 60 Tage Zeit, soweit es keine Nachfragen oder Probleme mit den Unterlagen gibt. Sollte während des Verfahrens eine Frage oder Aufforderung der Behörden aufkommen, sollten Antragssteller unbedingt auf gesetzte Fristen achten und zügig reagieren.
Genehmigung – ja oder nein
Das Ergebnis der Prüfung teilen die Mitgliedsstaaten schließlich wieder über das Portal CTIS dem Antragssteller mit.
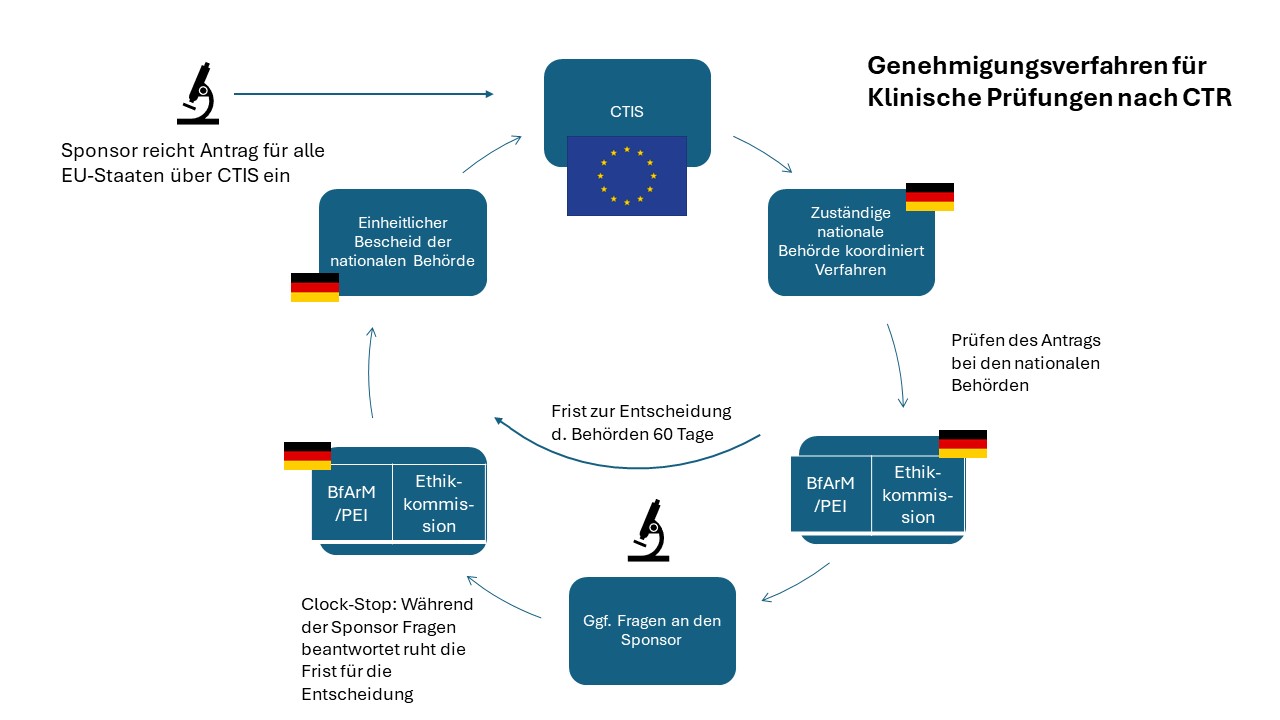 Das Genehmigungsverfahren nach der Clinical Trials Regulation
© Anja Segschneider
Das Genehmigungsverfahren nach der Clinical Trials Regulation
© Anja Segschneider
Pläne in Deutschland: Die nationale Pharmastrategie und klinische Prüfungen
Auch bei Klinischen Prüfungen, die ausschließlich in Deutschland durchgeführt werden, gelten die Regeln der CTR. Die Einreichung der Unterlagen erfolgt über das EU-Portal CTIS. Ein gewisser Gestaltungsspielraum der nationalen Rahmenbedingungen bleibt den einzelnen Staaten jedoch.
Diesen Gestaltungsspielraum möchte die Bundesregierung nun nutzen, um den Pharmastandort Deutschland im internationalen Vergleich attraktiver zu machen. Im Dezember 2023 veröffentlichte sie im Zuge der „nationalen Pharmastrategie“ daher das Strategiepapier „Verbesserung der Rahmenbedingungen für den Pharmabereich in Deutschland“ 10). Zu den Plänen gehört der Entwurf für ein „Medizinforschungsgesetz“, den Bundesgesundheitsminister Karl Lauterbach am 1. Dezember 2023 vorstellte und der Ende März 2024 vom Bundeskabinett beschlossen wurde.
Vorgesehen sind unter anderem:
Bündelung beim BfArM: Das BfArM soll zentraler Ansprechpartner für die Zulassung und Klinische Prüfung von Arzneimitteln, außer Impfstoffen und Blutprodukten, werden. Es soll außerdem eine interdisziplinär zusammengesetzte Bundes-Ethik-Kommission beim BfArM eingerichtet werden.
Kritik und Zukunft der nationalen Pharmastrategie
Die Pläne der nationalen Pharmastrategie blieben nicht ohne Kritik, wie unter anderem tagesschau.de11) berichtete. Insbesondere die vorgesehenen vertraulichen Erstattungsbeträge, die Pharmafirmen erlauben, ihre Preisgestaltung nicht mehr offen zu legen, sowie die geplante spezialisierte Ethikkommission wurden vor allem von den Krankenkassen sowie Ärztekammern bemängelt.
Bevor die Pläne verbindlich werden, müssen sie erst noch das weitere Gesetzgebungsverfahren durchlaufen. Dies könnte jedoch zeitnah erfolgen. Für das Genehmigungsverfahren von Klinischen Prüfungen wird die CTR als EU-Verordnung jedoch auch dann maßgeblich bleiben, wenn die nationale Pharmastrategie in Deutschland verabschiedet wird.