Umweltrisikobewertung von Humanarzneimitteln
Damit Medikamente nicht zum Risiko für die Umwelt werden
Täglich gelangen in Deutschland pharmazeutische Wirkstoffe in die Umwelt, mit potenziell schwerwiegenden Folgen für Ökosysteme und die menschliche Gesundheit. Die Umweltrisikobewertung (Environmental Risk Assessment, ERA) ist daher ein wesentlicher Bestandteil des Zulassungsverfahrens für Arzneimittel in Europa, um diese Risiken zu bewerten. Dr. Susanne Schwonbeck vom Fraunhofer-Institut für Toxikologie und Experimentelle Medizin ITEM gibt einen Einblick in die Herausforderungen und Lösungen der Umweltrisikobewertung.
Arzneimittelrückstände gelangen täglich in die Umwelt, sei es durch Ausscheidung nach der Einnahme oder unsachgemäße Entsorgung. 414 verschiedene Wirkstoffe sind laut Datenbank des Umweltbundesamtes (UBA) allein in Deutschland in der Umwelt zu finden, weltweit sind es 992 verschiedene pharmazeutische Wirkstoffe, die zum Teil in hohen Konzentrationen in Gewässern oder Böden vorkommen. Von dort aus können sie zur Gefahr werden: Sie schädigen Mikroorganismen, wodurch zum Beispiel Kläranlagen weniger wirksam werden, tragen zu Antibiotikaresistenzen bei und können sogar über das Trinkwasser zurück in die Bevölkerung gelangen.
Um diese Gefahren zu beherrschen, ist in Europa für jede Arzneimittelzulassung eine Umweltrisikobewertung, auch Environmental Risk Assessment (ERA) genannt, gesetzlich vorgeschrieben. Dabei wird geprüft, inwieweit ein Arzneimittel ein Risiko für die Umwelt darstellt und wie diese möglichen Risiken minimiert werden können. Wie die ERA abläuft, was Arzneimittelrückstände für die Umwelt bedeuten und wie die Rechtslage aussieht, erläuterte Dr. Susanne Schwonbeck vom Fraunhofer-Institut für Toxikologie und Experimentelle Medizin ITEM.
Wie Pharmazeutika in die Umwelt gelangen – und was sie dort anrichten können
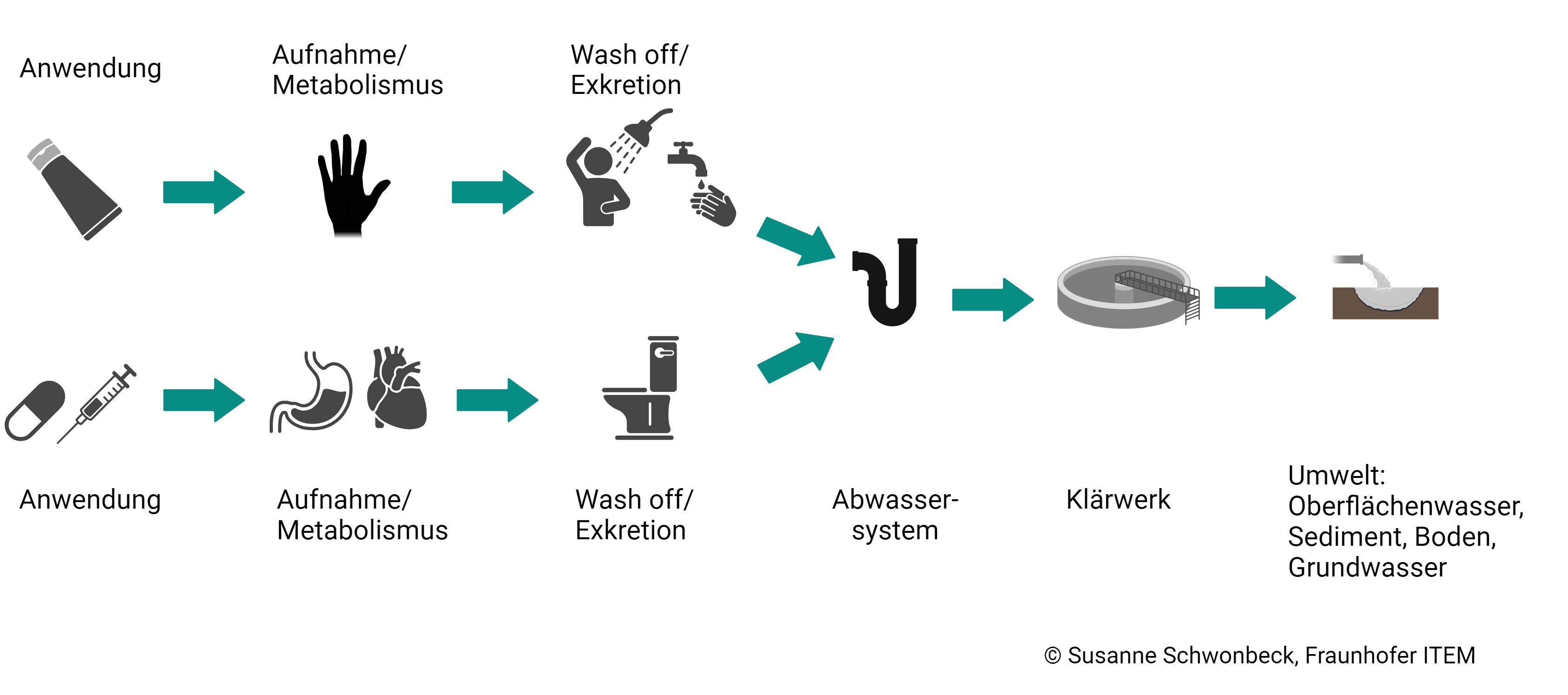 Medikamentenreste können auf verschiedenen Wegen in die Umwelt gelangen. © Schwonbeck – Fraunhofer ITEM
Medikamentenreste können auf verschiedenen Wegen in die Umwelt gelangen. © Schwonbeck – Fraunhofer ITEMDie Folgen von Arzneimitteln in der Umwelt sind oft nicht auf den ersten Blick absehbar. Denn nicht immer sind es die direkten Auswirkungen, wie etwa Rückstände von Hormonpräparaten im Trinkwasser, die am Ende schädliche Konsequenzen haben. Manchmal ist es eine ganze Kette von Ereignissen, wie das Geiersterben in Indien in den 1980er Jahren eindrucksvoll zeigt. Dort wurde das Medikament Diclofenac an Rinder verabreicht, deren Fleisch wiederum von Geiern gefressen wurde. 99,95 Prozent der indischen Geier starben in der Folge, was zu einer Zunahme von Krankheitsüberträgern wie Ratten und wilden Hunden führte. Eine Gefahr für den Menschen im Dominoeffekt.
„Es geht bei der Umweltrisikobewertung nicht nur um den Erhalt einer lebenswerten und intakten Umwelt, sondern auch um den Schutz des Menschen“, betont daher Dr. Susanne Schwonbeck vom Fraunhofer-Institut ITEM. Die Wissenschaftlerin befasst sich seit Jahren mit dem Thema Umweltrisikobewertung und berät auch Unternehmen hierzu. Als Ursachen für Arzneimittelrückstände in der Umwelt nennt sie sowohl Erzeugung, Nutzung als auch Entsorgung. Bei der Produktion von Pharmazeutika, die überwiegend außerhalb Europas stattfindet, geraten Wirkstoffe beispielsweise durch Abwässer in die Umwelt. Dasselbe geschieht, wenn sie von Menschen eingenommen oder Tieren verabreicht und später wieder ausgeschieden werden. Und auch bei der unsachgemäßen Entsorgung von Medikamenten können große Mengen an Arzneimittelrückständen freigesetzt werden. „Das Schlimmste, was Sie tun können, ist, Medikamente die Toilette hinunterzuspülen“, sagt Schwonbeck. „Am sichersten ist es, sie über den Hausmüll zu entsorgen. Der wird später verbrannt.“ Auch die Abgabe in der Apotheke sei eine gute Möglichkeit.
Dass Medikamentenreste in der Umwelt landen, lässt sich aber in der Praxis schlicht nicht vermeiden. Genau aus diesem Grund wird bei einer Umweltrisikobewertung festgestellt, was die Auswirkungen sind, wenn das passiert. „Bei der Umweltrisikobewertung werden nicht nur Risiken ermittelt, sondern anschließend Risikominderungsmaßnahmen durch die Behörden festgelegt. Es gibt dann auch eine entsprechende Kennzeichnung auf dem Beipackzettel“, erklärt Schwonbeck.
Rechtslage zur Umweltrisikobewertung
In der EU muss vor jeder Neuzulassung eine Umweltrisikobewertung durchgeführt werden. Dies gilt auch für Generika, Hybridprodukte und Kombinationsprodukte. Rechtsgrundlage ist vor allem die EU-Richtlinie 2001/83/EG zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel, die in Deutschland unter anderem im Arzneimittelgesetz (AMG) umgesetzt wurde. Die einschlägigen Vorschriften verlangen, dass Hersteller ermitteln, welche Risiken entstehen können, wenn ihre Wirkstoffe in die Umwelt gelangen, und wie diese gegebenenfalls minimiert werden können. „Dabei ist es wichtig, zwischen Risiko und Gefahr zu unterscheiden“, erläutert Schwonbeck. „Eine Gefahr ist das Potenzial eines Stoffes, Schäden zu verursachen, und ein Risiko ist die Wahrscheinlichkeit, dass diese Schäden tatsächlich eintreten.“ Diese beiden Aspekte müssen daher stets zusammen betrachtet werden. Eine Gefahr durch einen Wirkstoff ist zum Beispiel an sich noch nicht problematisch, solange das Risiko niedrig genug ist, dass der Schaden tatsächlich auch eintritt.
Der Ablauf der Umweltrisikobewertung ist in der technischen Leitlinie `Guideline on the environmental risk assessment of medicinal products for human use - Revision 1´1) festgelegt. Er erfolgt in verschiedenen Schritten, die von initialen Screening-Tests bis hin zu detaillierten experimentellen Studien reichen, je nachdem, ob ein potenzielles Risiko identifiziert wird.
Phase I beginnt mit der Berechnung der vorhergesagten Umweltkonzentration (PEC) eines Wirkstoffs. Überschreitet dieser Wert den Schwellenwert von 0,01 Mikrogramm pro Liter oder einen bestimmten Verteilungskoeffizienten, wird die nächste Phase eingeleitet. In dieser Phase werden insbesondere die Persistenz, Bioakkumulation und Toxizität (PBT) gescreent. Wenn die Grenzwerte nicht überschritten werden, endet das Verfahren bereits hier.
Es gibt jedoch auch Wirkstoffe, die bereits in sehr geringen Mengen ein Risiko darstellen. Hierzu gehören beispielsweise Hormone. Bei diesen Stoffen wird auch unterhalb des Grenzwertes die Phase II durchgeführt.
Phase II gliedert sich in die Unterphasen 2A und 2B und umfasst experimentelle Untersuchungen zur Toxizität auf verschiedene Umweltorganismen wie Algen, Kleinkrebse, Fische und Mikroorganismen. Zusätzlich werden auch die Abbaubarkeit des Wirkstoffs und sein Verhalten in der Umwelt, einschließlich der Absorption und möglicher chemischer Umwandlungen, betrachtet. Die Ergebnisse dieser Studien bilden die Grundlage für die Berechnung des Risikos, das von einem bestimmten Arzneimittel ausgeht.
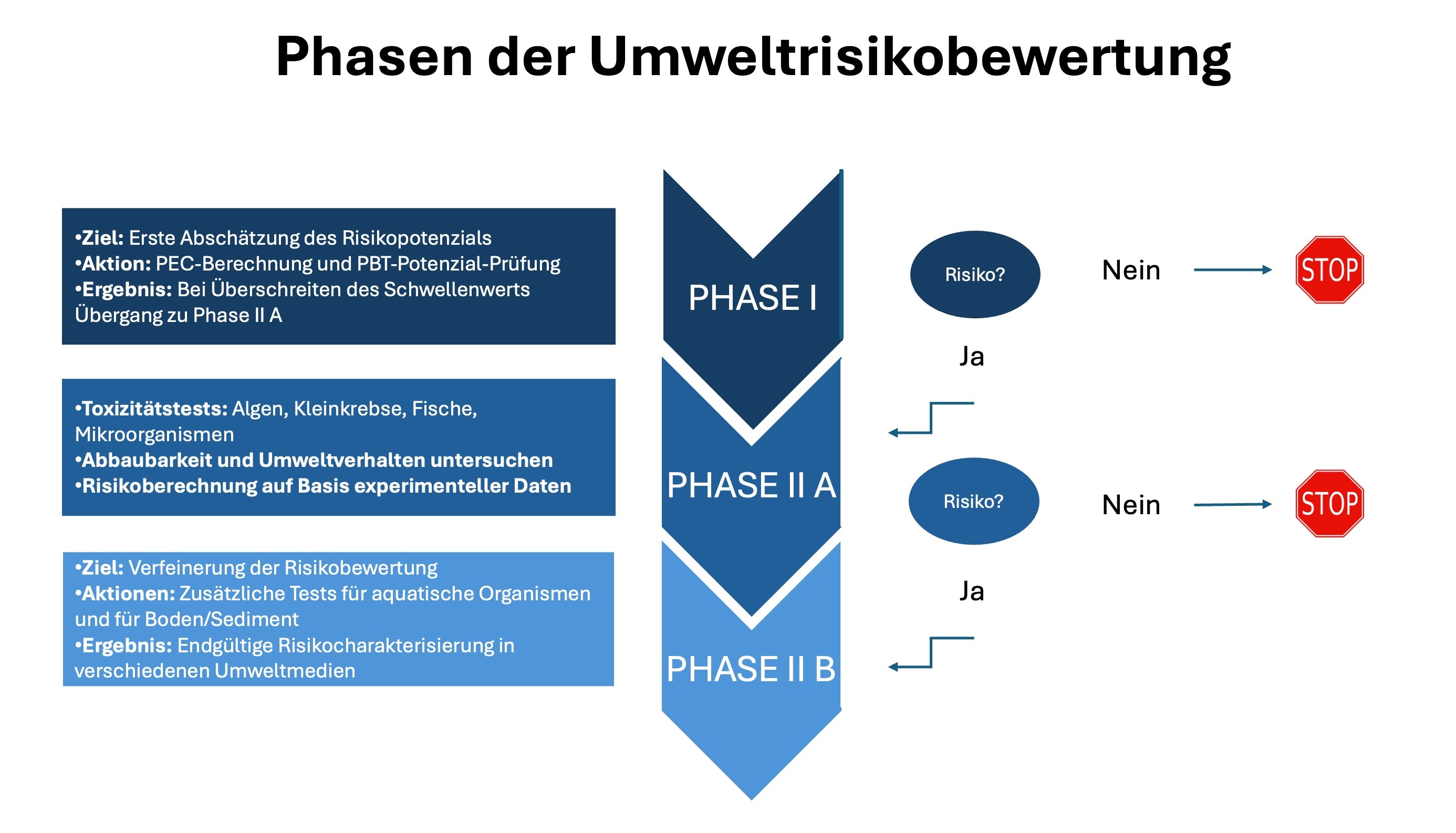 Die verschiedenen Phasen einer Umweltrisikobewertung. © Anja Segschneider (in Anlehnung an Schwonbeck – Fraunhofer ITEM)
Die verschiedenen Phasen einer Umweltrisikobewertung. © Anja Segschneider (in Anlehnung an Schwonbeck – Fraunhofer ITEM)Die komplette Untersuchung der Gefahren und Risiken muss am Ende mit dem Zulassungsdossier beim Antrag der Zulassung des Arzneimittels eingereicht werden. Die Zusammenfassung wird von den Behörden in sogenannten Assessment Reports (Bewertungsberichte) veröffentlicht. „Gegebenenfalls gibt es dann noch Auflagen, weitere Studien durchzuführen“, sagt Schwonbeck. „Zur Versagung der Zulassung führen die Ergebnisse der Umweltrisikobewertung am Ende jedoch nicht.“
Umweltrisikobewertung: Offene Fragen
Zwar bietet die Umweltrisikobewertung eine Möglichkeit, pharmazeutische Stoffe im Auge zu behalten, lückenlos funktioniert sie jedoch nicht. „Letztendlich sind mehr als 2.500 verschiedene Humanarzneimittelwirkstoffe in Deutschland zugelassen und davon haben ein bisschen weniger als die Hälfte tatsächlich auch eine Umweltrelevanz“, sagt Schwonbeck. „Wie viel tatsächlich pro Jahr verbraucht wird und damit auch in die Umwelt gelangt, ist jedoch schwer zu sagen. Es gibt solche Daten für Antibiotika in der Tiermast. Bei Humanarzneien wird dies jedoch nicht erfasst.“ Zudem tauchten in Klärwerken und Trinkwasserbetrieben immer wieder Wirkstoffe oder Abbauprodukte auf, die sich nicht bewerten ließen. Es fehlten die Daten für eine Beurteilung. Woher diese stammen? Möglicherweise handelt es sich um alte Wirkstoffe, die noch nicht auf ihre Umweltverträglichkeit getestet wurden. Schwonbeck bemängelt in diesem Zusammenhang auch fehlende Daten und die schlechte Verfügbarkeit bereits vorhandener Umweltdaten, die die Forschung erschwere. Es müsse zumindest europaweit einen besseren Austausch geben. „Zum jetzigen Zeitpunkt wird intensiv an der Pharmagesetzgebung gearbeitet. Ich denke, dort wird sich hoffentlich auch bei der Umweltrisikobewertung noch etwas tun.“